Journal of Theoretical & Computational Science
Open Access
ISSN: 2376-130X
ISSN: 2376-130X
Research Article - (2014) Volume 1, Issue 2
the present methodical study, FT-IR, FT-Raman and NMR spectra of the L-Valine are recorded and the observed vibrational frequencies are assigned. The hybrid computational calculations are carried out by HF and DFT (B3LYP and B3PW91) methods with 6-31+G(d,p) and 6-311++G(d,p) basis sets and the corresponding results are tabulated. The alternation of structure of amino acid due to the subsequent substitutions of CH3 is investigated. The vibrational sequence pattern of the molecule related to the zwitter ion motion is analyzed. Moreover, 13C NMR and 1H NMR are calculated by using the gauge independent atomic orbital (GIAO) method with B3LYP methods and the 6-311++G(d,p) basis set and their spectra are simulated and the chemical shifts related to TMS are compared. A study on the electronic and optical properties; absorption wavelengths, excitation energy, dipole moment and frontier molecular orbital energies, are performed by HF and DFT methods. The calculated HOMO and LUMO energies and the kubo gap analysis show that the occurring of charge transformation within the molecule. Besides frontier molecular orbitals (FMO), molecular electrostatic potential (MEP) was performed. NLO properties related to Polarizability and hyperpolarizability is also discussed.
<Keywords: L-Valine, Optical properties, Gauge independent atomic orbital, Chemical shifts, FMO
L-Valine is a non polar α amino acid also named as 2-amino-3- methylbutanoic acid which is one of 20 proteinnogenic amino acids [1]. L-Valine is an essential amino acid; hence it must be ingested, usually as a component of proteins. It is necessary for smooth nervous system and cognitive functioning. It is one of the three Branched Chain Amino Acids (BCAAs), the other two being L-leucine and L-isoleucine.
L-Valine also greatly benefits the regulation of the immune system. L-Valine is essential for muscle tissue repair and muscle metabolism, and also increases exercise endurance. L-Valine also greatly benefits the regulation of the immune system. Perhaps the biggest benefits are experienced by athletes who perform long distance sports and bodybuilding. It is synthesized in plants via several steps starting from pyruvic acid. The initial part of the pathway also leads to leucine. The intermediate α-ketoisovalerate undergoes reductive amination with glutamate [2].
L-Valine is also aliphatic nonpolar chain having both a primary amino group and a carboxyl group in which the proton is exchanged between them. So the amino acid exists as zwitterions [3]. In this respect, the resources with amino acids are interesting materials for NLO applications. The amino acids contain asymmetric carbon atoms which make it optically active and most of them crystallize in non centrosymmetric space groups. Bulk crystals of amino acids have physical properties, which makes them ideal candidates for nonlinear optical devices. In particular, optically amino acids posses’ wide optical transparency ranges in UV-Visible spectral region, favorable hardness due to their zwitterionic nature and large hyperpolarizability [4-6]. In order to establish the electrical and optical properties of the L-Valine, it is very important that, the vibrational, magnetic resonance and UVVisible spectroscopic study to be carried out. There were some previous works have been reported [7,8] on L-Valine complexes using FT-IR and FT-Raman spectral studies. In this work, the FT-IR, FT-Raman, 1H and 13C NMR, Frontier molecular studies and Kubo gap analysis have been carried out on L-Valine.
The compound L-Valine is purchased from Sigma–Aldrich Chemicals, USA, which is of spectroscopic grade and hence used for recording the spectra as such without any further purification. The FT-IR spectrum of the compound is recorded in Bruker IFS 66V spectrometer in the range of 4000–400 cm−1. The spectral resolution is ± 2 cm−1. The FT-Raman spectrum of L-Valine is also recorded in the same instrument with FRA 106 Raman module equipped with Nd:YAG laser source operating at 1.064 μm line widths with 200 mW power. The spectra are recorded in the range of 4000–100 cm−1 with scanning speed of 30 cm−1 min−1 of spectral width 2 cm−1. The frequencies of all sharp bands are accurate to ±1 cm−1. The 13C NMR spectrum is recorded by Spin solve high resolution bench top FTNMR Spectrometer. The operating frequency: 42.5 MHz Proton with Resolution: 50% line width, <25 ppb (1 Hz) in Sensitivity is greater than 10000:1.
In the present work, HF and some of the hybrid methods; B3LYP and B3PW91 are carried out using the basis sets 6-31+G(d,p) and 6-311+G(d,p). All these calculations are performed using GAUSSIAN 09W [9] program package on Pentium IV processor in personal computer. In DFT methods; Becke’s three parameter hybrids function combined with the Lee-Yang-Parr correlation function (B3LYP) [10,11], Becke’s three parameter exact exchange-function (B3) [12] combined with gradient-corrected correlational functional of Lee, Yang and Parr (LYP) [13,14] and Perdew and Wang (PW91) [15,16] predict the best results for molecular geometry and vibrational frequencies for moderately larger molecules. The calculated frequencies are scaled down to yield the coherent with the observed frequencies. The scaling factors are 0.904 for HF/6-311++G(d,p) method. For B3LYP/6-31+/6- 311++G(d,p) basis set, the scaling factors are 0.984, 0.973, 0.943 and 1.02/0.988, 0.946 and 0.960. For B3PW91/6-31+G/6-311+G(d,p) basis set, the scaling factors are 0.975,0.982, 0.932 and 1.02/0.980, 0.945, 0.970 and 1.02. The optimized molecular structure of the molecule is obtained from Gaussian 09 and Gauss view program and is shown in Figure 1. The comparative optimized structural parameters such as bond length, bond angle and dihedral angle are presented in Table 1. The observed (FT-IR and FT-Raman) and calculated vibrational frequencies and vibrational assignments are submitted in Table 2. Experimental and simulated spectra of IR and Raman are presented in the Figures 2 and 3, respectively.

Figure 1: Molecular Structure of L-Valine.

Figure 2: Experimental [A] and calculated [B, C & D] FT-IR spectra of L-Valine.

Figure 3: Experimental [A] and calculated [B, C & D] FT-Raman spectra of L-Valine.
| Geometrical Parameters | Methods | ||||||
|---|---|---|---|---|---|---|---|
| HF | B3LYP | B3PW91 | |||||
| 6-311+G (d, p) | 6-31+G (d, p) | 6-311+G (d, p) | 6-31+G (d, p) | 6-311+G (d, p) | |||
| Bond length (Å) | |||||||
| O1-C3 | 1.321 | 1.343 | 1.342 | 1.337 | 1.336 | ||
| O2-C3 | 1.179 | 1.210 | 1.202 | 1.209 | 1.201 | ||
| C3-C4 | 1.537 | 1.553 | 1.553 | 1.549 | 1.538 | ||
| C4-H5 | 1.089 | 1.099 | 1.097 | 1.100 | 1.097 | ||
| C4-N6 | 1.461 | 1.478 | 1.477 | 1.471 | 1.468 | ||
| C4-C10 | 1.544 | 1.553 | 1.552 | 1.547 | 1.548 | ||
| C6-H7 | 1.001 | 1.017 | 1.016 | 1.016 | 1.012 | ||
| C6-H8 | 2.032 | 1.867 | 1.881 | 1.825 | 1.884 | ||
| C6-H9 | 0.997 | 1.014 | 1.012 | 1.013 | 1.013 | ||
| C10-H11 | 1.088 | 1.100 | 1.097 | 1.100 | 1.099 | ||
| C10-C12 | 1.533 | 1.539 | 1.537 | 1.533 | 1.520 | ||
| C10-C16 | 1.533 | 1.538 | 1.536 | 1.532 | 1.532 | ||
| C12-H13 | 1.079 | 1.089 | 1.087 | 1.090 | 1.090 | ||
| C12-H14 | 1.085 | 1.095 | 1.093 | 1.095 | 1.093 | ||
| C12-H15 | 1.088 | 1.098 | 1.096 | 1.097 | 1.094 | ||
| C16-H17 | 1.084 | 1.094 | 1.092 | 1.094 | 1.094 | ||
| C16-H18 | 1.088 | 1.098 | 1.096 | 1.097 | 1.095 | ||
| C16-H19 | 1.085 | 1.094 | 1.092 | 1.094 | 1.093 | ||
| Bond angle (°) | |||||||
| O1-C3-O2 | 121.59 | 122.03 | 122.05 | 122.38 | 122.98 | ||
| O1-C3-C4 | 113.96 | 113.31 | 113.30 | 112.96 | 113.45 | ||
| O2-C3-C4 | 124.41 | 124.62 | 124.61 | 124.62 | 123.54 | ||
| C3-C4-H5 | 104.64 | 104.97 | 104.75 | 104.89 | 106.34 | ||
| C3-C4-N6 | 107.31 | 107.13 | 107.38 | 107.03 | 107.66 | ||
| C3-C4-C10 | 113.01 | 113.61 | 113.47 | 113.69 | 111.27 | ||
| H5-C4-N6 | 106.01 | 106.21 | 106.18 | 106.44 | 112.99 | ||
| H5-C4-C10 | 108.20 | 107.86 | 107.87 | 107.83 | 108.21 | ||
| N6-C4-C10 | 116.76 | 116.21 | 116.32 | 116.12 | 110.30 | ||
| C4-N6-H7 | 111.54 | 111.49 | 111.47 | 111.41 | 112.12 | ||
| C4-N6-H8 | 81.09 | 84.62 | 84.15 | 84.86 | 83.18 | ||
| C4-N6-H9 | 111.64 | 112.00 | 111.89 | 112.11 | 111.80 | ||
| H7-N6-H8 | 108.16 | 106.20 | 106.43 | 106.36 | 131.59 | ||
| H7-N6-H9 | 107.64 | 107.52 | 107.46 | 107.58 | 107.52 | ||
| H8-C6-H9 | 133.48 | 132.68 | 132.99 | 132.22 | 108.13 | ||
| C4-C10-H11 | 107.78 | 107.50 | 107.50 | 107.53 | 106.74 | ||
| C4-C10-C12 | 111.42 | 111.75 | 111.70 | 111.71 | 112.86 | ||
| C4-C10-C16 | 111.55 | 111.3 | 111.40 | 111.19 | 110.75 | ||
| H11-C10-C12 | 108.19 | 108.22 | 108.23 | 108.25 | 108.31 | ||
| H11-C10-C16 | 108.33 | 108.34 | 108.33 | 108.43 | 108.39 | ||
| C12-C10-C16 | 109.43 | 109.55 | 109.53 | 109.58 | 109.60 | ||
| C10-C12-H13 | 111.76 | 111.61 | 111.66 | 111.53 | 112.50 | ||
| C10-C12-H14 | 110.10 | 110.06 | 110.09 | 110.13 | 109.59 | ||
| C10-C12-H15 | 110.45 | 110.39 | 110.37 | 110.40 | 111.06 | ||
| H13-C12-H14 | 108.24 | 108.74 | 108.65 | 108.77 | 108.21 | ||
| H13-C12-H15 | 108.67 | 108.46 | 108.47 | 108.42 | 107.40 | ||
| H14-C12-H15 | 107.46 | 107.4 | 107.45 | 107.44 | 107.88 | ||
| C10-C16-H17 | 112.75 | 113.12 | 113.09 | 113.21 | 113.78 | ||
| C10-C16-H18 | 111.24 | 111.06 | 111.09 | 111.02 | 110.67 | ||
| C10-C16-H19 | 109.92 | 109.87 | 109.86 | 109.93 | 110.05 | ||
| H17-C16-H18 | 108.05 | 107.99 | 108.02 | 107.99 | 107.75 | ||
| H17-C16-H19 | 107.28 | 107.24 | 107.25 | 107.17 | 106.67 | ||
| H18-C16-H19 | 107.35 | 107.29 | 107.27 | 107.25 | 107.63 | ||
| Dihedral angles (˚) | |||||||
| O1-C3-C4-H5 | -79.63 | -93.09 | -92.17 | -94.43 | -145.33 | ||
| O1-C3-C4-N6 | 32.70 | 19.54 | 20.43 | 18.38 | -23.95 | ||
| O1-C3-C4-C10 | 162.86 | 149.28 | 150.41 | 147.98 | 97.03 | ||
| O2-C3-C4-H5 | 98.87 | 85.14 | 86.09 | 83.82 | 35.52 | ||
| O2-C3-C4-N6 | -148.78 | -162.21 | -161.29 | -163.35 | 156.90 | ||
| O2-C3-C4-C10 | -18.62 | -32.47 | -31.31 | -33.75 | -82.10 | ||
| C3-C4-N6-H7 | 80.52 | 88.46 | 87.97 | 89.56 | 151.93 | ||
| C3-C4-N6-H8 | -25.57 | -16.85 | -17.41 | -16.02 | 19.63 | ||
| O3-C4-C6-H9 | -158.98 | -150.99 | -151.64 | -149.80 | -87.21 | ||
| H5-C4-O6-H7 | -168.06 | -159.73 | -160.38 | -158.67 | -90.92 | ||
| H5-C4-O6-H8 | 85.83 | 94.94 | 94.22 | 95.73 | 136.76 | ||
| H5-C4-O6-H9 | -47.57 | -39.19 | -40.01 | -38.03 | 29.92 | ||
| C10-C4-N6-H7 | -47.48 | -39.77 | -40.38 | -38.64 | 30.34 | ||
| C10-C4-N6-H8 | -153.59 | -145.09 | -145.77 | -144.22 | -101.95 | ||
| C10-C4-N6-H9 | 72.99 | 80.76 | 79.99 | 81.99 | 151.19 | ||
| C3-C4-C10-H11 | -47.10 | -49.82 | -49.48 | -50.17 | -67.78 | ||
| C3-C4-C10-C12 | 71.45 | 68.79 | 69.11 | 68.46 | 51.10 | ||
| C3-C4-C10-C16 | -165.9 | -168.34 | -168.03 | -168.74 | 174.41 | ||
| C5-C4-C10-H11 | -162.49 | -165.75 | -165.05 | -166.03 | 175.72 | ||
| C5-C4-C10-C12 | -43.94 | -47.14 | -46.46 | -47.39 | -65.38 | ||
| C5-C4-C10-C16 | 78.69 | 75.72 | 76.39 | 75.39 | 57.92 | ||
| N6-C4-C10-H11 | 78.08 | 75.17 | 75.85 | 74.68 | 51.64 | ||
| N6-C4-C10-C12 | -163.35 | -166.20 | -165.55 | -166.66 | 170.52 | ||
| N6-C4-C10-C16 | -40.71 | -43.34 | -42.69 | -43.88 | -66.15 | ||
| C4-C10-C12-H13 | -57.57 | -58.20 | -58.30 | -57.86 | -60.55 | ||
| C4-C10-C12-H14 | -177.91 | -179.05 | -179.09 | -178.74 | 179.0 | ||
| C4-C10-C12-H15 | 63.55 | 62.50 | 62.44 | 62.76 | 59.88 | ||
| H11-C10-C12-H13 | 60.73 | 59.97 | 59.85 | 60.35 | 57.41 | ||
| H11-C10-C12-H14 | -59.59 | -60.86 | -60.93 | -60.52 | -63.03 | ||
| H11-C10-C12-H15 | -178.13 | -179.31 | -179.39 | -179.01 | 177.85 | ||
| C16-C10-C12-H13 | 178.58 | 177.91 | 177.78 | 178.44 | 175.50 | ||
| C16-C10-C12-H14 | 58.24 | 57.06 | 56.99 | 57.55 | 55.05 | ||
| C16-C10-C12-H15 | -60.28 | -61.37 | -61.46 | -60.93 | -64.05 | ||
| C4-C10-C16-H17 | 63.43 | 62.42 | 62.32 | 62.43 | 59.27 | ||
| C4-C10-C16-H18 | -58.14 | -59.21 | -59.33 | -59.23 | -62.22 | ||
| C4-C10-C16-H19 | -176.91 | -177.75 | -177.87 | -177.74 | 178.92 | ||
| H11-C10-C16-H17 | -55.04 | -55.59 | -55.71 | -55.59 | -57.51 | ||
| H11-C10-C16-H18 | -176.61 | -177.22 | -177.38 | -177.25 | -179.02 | ||
| H11-C10-C16-H19 | 64.60 | 64.22 | 64.07 | 64.22 | 62.13 | ||
| C12-C10-C16-H17 | -172.79 | -173.45 | -173.58 | -173.56 | -175.55 | ||
| C12-C10-C16-H18 | 65.62 | 64.90 | 64.75 | 64.76 | 62.94 | ||
| C12-C10-C16-H19 | -53.14 | -53.64 | -53.78 | -53.74 | -55.904 | ||
Table 1: Optimized geometrical parameters for L-Valine computed at HF/DFT(B3LYP&B3PW91) with 6-31& 6-311G(d, p) basis sets.
| S. No | Symmetry Species Cs | Observed Frequency(cm-1) FTIR FTRaman | Methods | Vibrational Assignments | |||||
|---|---|---|---|---|---|---|---|---|---|
| HF | B3LYP | B3PW91 | |||||||
| 6-311+G (d, p) | 6-31+G (d, p) | 6-311+G (d, p) | 6-31+G (d, p) | 6-311+G (d, p) | |||||
| 1 | A′ | 3550w | - | 3662 | 3551 | 3550 | 3547 | 3551 | (O-H) υ |
| 2 | A′ | 3460s | 3460m | 3458 | 3455 | 3462 | 3472 | 3470 | (N-H) υ |
| 3 | A′ | 3350s | 3350m | 3373 | 3351 | 3347 | 3348 | 3340 | (N-H) υ |
| 4 | A′ | 2970s | 2970vs | 2976 | 2986 | 2967 | 2965 | 2970 | (C-H) υ |
| 5 | A′ | 2930vs | - | 2922 | 2934 | 2918 | 2917 | 2941 | (C-H) υ |
| 6 | A′ | 2920vs | 2920s | 2911 | 2922 | 2906 | 2906 | 2936 | (C-H) υ |
| 7 | A′ | - | 2910vs | 2900 | 2915 | 2898 | 2900 | 2925 | (C-H) υ |
| 8 | A′ | 2890s | 2890s | 2868 | 2863 | 2903 | 2840 | 2877 | (C-H) υ |
| 9 | A′ | 2880s | - | 2855 | 2856 | 2895 | 2834 | 2871 | (C-H) υ |
| 10 | A′ | 2860s | - | 2851 | 2854 | 2851 | 2832 | 2862 | (C-H) υ |
| 11 | A′ | 2850s | - | 2846 | 2841 | 2837 | 2820 | 2844 | (C-H) υ |
| 12 | A′ | 1710m | - | 1834 | 1732 | 1727 | 1731 | 1727 | (C=O) υ |
| 13 | A′ | 1620vs | 1620w | 1629 | 1618 | 1619 | 1620 | 1620 | (O-H) δ |
| 14 | A′ | - | 1495s | 1483 | 1486 | 1483 | 1494 | 1503 | (N-H) δ |
| 15 | A′ | 1465vs | - | 1466 | 1468 | 1467 | 1466 | 1470 | (N-H) δ |
| 16 | A′ | - | 1460s | 1462 | 1460 | 1459 | 1457 | 1456 | (CH3) α |
| 17 | A′ | - | 1450vs | 1452 | 1452 | 1451 | 1449 | 1446 | (CH3)α |
| 18 | A′ | 1400vs | - | 1401 | 1405 | 1405 | 1404 | 1395 | (CH3) α |
| 19 | A′ | 1395vs | - | 1397 | 1388 | 1394 | 1394 | 1393 | (CH3) α |
| 20 | A″ | 1375vs | - | 1380 | 1368 | 1383 | 1374 | 1374 | (O-H) γ |
| 21 | A′ | 1370vs | - | 1371 | 1366 | 1372 | 1361 | 1373 | (C-N) υ |
| 22 | A′ | 1350vs | 1350m | 1352 | 1348 | 1353 | 1343 | 1347 | (C-H) δ |
| 23 | A′ | 1325s | - | 1314 | 1337 | 1320 | 1334 | 1323 | (C-H) δ |
| 24 | A′ | 1270s | 1270w | 1267 | 1277 | 1262 | 1272 | 1279 | (C-O) υ |
| 25 | A′ | 1190m | - | 1190 | 1188 | 1191 | 1197 | 1181 | (C-H) δ |
| 26 | A′ | - | 1170m | 1179 | 1171 | 1180 | 1180 | 1171 | (C-H) δ |
| 27 | A′ | 1165m | - | 1163 | 1165 | 1167 | 1164 | 1161 | (C-H) δ |
| 28 | A′ | 1140m | 1140m | 1126 | 1145 | 1130 | 1133 | 1141 | (C-H) δ |
| 29 | A′ | 1105vw | - | 1092 | 1106 | 1105 | 1115 | 1115 | (C-H) δ |
| 30 | A′ | - | 1050vs | 1041 | 1044 | 1047 | 1052 | 1054 | (C-C) υ |
| 31 | A′ | 940m | 940s | 931 | 945 | 939 | 932 | 930 | (C-C-N) υ |
| 32 | A′ | - | 935s | 937 | 940 | 953 | 943 | 933 | C-(CH3) υ |
| 33 | A″ | 900w | - | 908 | 911 | 899 | 924 | 902 | (C-C) υ (N-H) γ |
| 34 | A″ | - | 870vs | 885 | 864 | 869 | 868 | 869 | (N-H) γ |
| 35 | A″ | 850w | - | 842 | 838 | 840 | 846 | 852 | (C-H) γ |
| 36 | A″ | - | 820m | 811 | 792 | 805 | 832 | 828 | (C-H) γ |
| 37 | A″ | 720s | - | 727 | 774 | 776 | 773 | 767 | (C-H) γ |
| 38 | A″ | 710s | 697 | 705 | 720 | 713 | 708 | (C-H) γ | |
| 39 | A″ | 560s | 560w | 589 | 564 | 552 | 567 | 628 | (C-H) γ |
| 40 | A″ | - | 500w | 512 | 541 | 518 | 542 | 511 | (C-H) γ |
| 41 | A″ | 450m | - | 450 | 450 | 431 | 449 | 443 | (C-H) γ |
| 42 | A″ | - | 390w | 395 | 384 | 386 | 383 | 387 | (C-C) δ (C-H) γ |
| 43 | A′ | - | 360w | 365 | 360 | 361 | 364 | 363 | (C-C) δ |
| 44 | A″ | - | 340w | 339 | 341 | 340 | 332 | 337 | (CO2) γ |
| 45 | A′ | - | 310w | 325 | 329 | 324 | 325 | 316 | C-(CH3) δ |
| 46 | A″ | - | 280w | 278 | 273 | 273 | 286 | 269 | (C-C) γ (C-N) γ |
| 47 | A″ | - | 220w | 249 | 234 | 234 | 232 | 221 | C-(CH3) γ |
| 48 | A″ | - | 210w | 209 | 216 | 215 | 215 | 199 | (CH3) τ |
| 49 | A″ | - | 200w | 195 | 202 | 191 | 202 | 185 | (CH3) τ |
| 50 | A″ | - | 150w | 74 | 78 | 70 | 79 | 74 | (NH2) τ |
| 51 | A″ | - | 140w | 60 | 41 | 41 | 41 | 39 | (C=O-OH) τ |
VS –Very strong; S – Strong; m- Medium; w – weak; as - Asymmetric; s – symmetric; υ – stretching; α –deformation, δ - In plane bending; γ - out plane bending; τ – Twisting
Table 2: Observed and HF and DFT (B3LYP & B3PW91) with 6-31+G(d,p) & 6-311+G (d,p) level.
The 1H and 13C NMR isotropic shielding are calculated with the GIAO method [17] using the optimized parameters obtained from B3LYP/6-311++G(d,p) method. 13C isotropic magnetic shielding (IMS) of any X carbon atoms is made according to value 13C IMS of TMS, CSX=IMSTMS-IMSx. The 1H and 13C isotropic chemical shifts of TMS at B3LYP methods with 6-311++G(d,p) level using the IEFPCM method in DMSO and CCl4. The absolute chemical shift is found between isotropic peaks and the peaks of TMS [18].
The electronic properties; HOMO-LUMO energies, absorption wavelengths and oscillator strengths are calculated using B3LYP method of the time-dependent DFT (TD-DFT) [19,20], basing on the optimized structure in gas phase and solvent[DMSO, ethanol, methanol and acetone] mixed phase. Thermodynamic properties of L-Valine at 298.15°C have been calculated in gas phase using B3LYP/6- 311++G(d,p) method. Moreover, the dipole moment, nonlinear optical (NLO) properties, linear polarizabilities and first hyperpolarizabilities and chemical hardness have also been studied.
Molecular geometry
The molecular structure of L-Valine belongs to CS point group symmetry. The optimized structure of the molecule is obtained from Gaussian 09 and Gauss view program [12] and is shown in Figure 1. The present molecule contains one amino and COOH group and two methyl groups.
The structure optimization and zero point vibrational energy of the compound in HF and DFT(B3LYP/B3PW91) with 6-31+/6-311+G(d,p) are 110.78, 103.52, 103.21, 103.91 and 103.41 Kcal/Mol, respectively. The calculated value of HF is greater than the values of DFT method because the assumption of ground state energy in HF is greater than the true energy. The breaking of L-Valine structure belongs to multiple planes which are due to the couple of amino, acid and methyl groups. The bond length between C3-C4 and C4-C10 are nearly equal since both C are balanced by NH2 and CH3 in the chain. The bond lengths of C10-C12 and C10-C16 are nearly equal due to the balance of CH3 groups in the chain. The entire C-H bonds in the chain and methyl group having almost equal inter nuclear distance. This view showed that there are no further substitutions in the chain. Form the optimized molecular structure; it is observed that there is no arithmetical change in the chain. So there is no further change in geometrical property.
Vibrational assignments
In order to obtain the spectroscopic signature of the L-Valine, the computational calculations are performed for frequency analysis. The molecule, has CS point group symmetry, consists of 19 atoms, so it has 51 normal vibrational modes. On the basis of CS symmetry, the 51 fundamental vibrations of the molecule can be distributed as 33 in-plane vibrations of A′ species and 18 out of plane vibrations of A″ species, i.e., Γvib=33 A′ + 18 A″. In the CS group symmetry of molecule is non-planar structure and has the 51 vibrational modes span in the irreducible representations.
The harmonic vibrational frequencies (unscaled and scaled) calculated at HF, B3LYP and B3PW91 levels using the triple split valence basis set along with the diffuse and polarization functions, 6-31+/6-311++G(d,p) and observed FT-IR and FT-Raman frequencies for various modes of vibrations have been presented in Tables 2 and 3. Comparison of frequencies calculated at HF and B3LYP/B3PW91 with the experimental values reveal the over estimation of the calculated vibrational modes due to the neglect of a harmonicity in real system. Inclusion of electron correlation in the density functional theory to certain extends makes the frequency values smaller in comparison with the HF frequency data. Reduction in the computed harmonic vibrations, although basis set sensitive is only marginal as observed in the DFT values using 6-311+G(d,p).
| S.No | Observed frequency | Calculated frequency | |||||
|---|---|---|---|---|---|---|---|
| HF | B3LYP | B3PW91 | |||||
| 6-311+G (d, p) | 6-31+G (d, p) | 6-311+G (d, p) | 6-31+G (d, p) | 6-311+G (d, p) | |||
| 1 | 3550 | 4051 | 3609 | 3593 | 3638 | 3623 | |
| 2 | 3460 | 3825 | 3511 | 3504 | 3536 | 3541 | |
| 3 | 3350 | 3731 | 3444 | 3472 | 3409 | 3461 | |
| 4 | 2970 | 3292 | 3166 | 3146 | 3181 | 3143 | |
| 5 | 2930 | 3232 | 3111 | 3094 | 3130 | 3112 | |
| 6 | 2920 | 3220 | 3099 | 3082 | 3118 | 3107 | |
| 7 | 2910 | 3208 | 3091 | 3073 | 3112 | 3095 | |
| 8 | 2890 | 3173 | 3036 | 3024 | 3047 | 3044 | |
| 9 | 2880 | 3158 | 3029 | 3016 | 3041 | 3038 | |
| 10 | 2860 | 3154 | 3027 | 3014 | 3039 | 3029 | |
| 11 | 2850 | 3148 | 3013 | 2999 | 3026 | 3010 | |
| 12 | 1710 | 2029 | 1837 | 1831 | 1857 | 1853 | |
| 13 | 1620 | 1802 | 1663 | 1660 | 1662 | 1653 | |
| 14 | 1495 | 1640 | 1527 | 1521 | 1521 | 1503 | |
| 15 | 1465 | 1622 | 1509 | 1505 | 1504 | 1500 | |
| 16 | 1460 | 1617 | 1501 | 1496 | 1494 | 1486 | |
| 17 | 1450 | 1606 | 1492 | 1488 | 1486 | 1475 | |
| 18 | 1400 | 1550 | 1428 | 1422 | 1430 | 1423 | |
| 19 | 1395 | 1545 | 1411 | 1411 | 1420 | 1421 | |
| 20 | 1375 | 1527 | 1406 | 1400 | 1399 | 1402 | |
| 21 | 1370 | 1517 | 1388 | 1389 | 1386 | 1373 | |
| 22 | 1350 | 1496 | 1370 | 1369 | 1368 | 1347 | |
| 23 | 1325 | 1454 | 1337 | 1336 | 1334 | 1323 | |
| 24 | 1270 | 1401 | 1277 | 1277 | 1272 | 1305 | |
| 25 | 1190 | 1316 | 1207 | 1205 | 1219 | 1250 | |
| 26 | 1170 | 1304 | 1203 | 1194 | 1210 | 1207 | |
| 27 | 1165 | 1286 | 1184 | 1181 | 1185 | 1185 | |
| 28 | 1140 | 1246 | 1145 | 1144 | 1154 | 1141 | |
| 29 | 1105 | 1208 | 1106 | 1105 | 1115 | 1081 | |
| 30 | 1050 | 1152 | 1061 | 1060 | 1071 | 1073 | |
| 31 | 995 | 1074 | 985 | 981 | 992 | 970 | |
| 32 | 940 | 1036 | 966 | 965 | 967 | 952 | |
| 33 | 900 | 1004 | 936 | 936 | 941 | 930 | |
| 34 | 870 | 979 | 916 | 905 | 931 | 920 | |
| 35 | 850 | 931 | 889 | 875 | 908 | 878 | |
| 36 | 820 | 897 | 840 | 839 | 847 | 854 | |
| 37 | 720 | 804 | 823 | 820 | 829 | 825 | |
| 38 | 710 | 771 | 725 | 729 | 731 | 722 | |
| 39 | 560 | 651 | 580 | 584 | 582 | 675 | |
| 40 | 500 | 566 | 550 | 548 | 556 | 549 | |
| 41 | 450 | 498 | 457 | 456 | 457 | 439 | |
| 42 | 390 | 437 | 407 | 408 | 411 | 387 | |
| 43 | 360 | 404 | 360 | 361 | 364 | 374 | |
| 44 | 340 | 375 | 350 | 349 | 356 | 337 | |
| 45 | 310 | 360 | 349 | 342 | 349 | 316 | |
| 46 | 280 | 308 | 290 | 289 | 291 | 274 | |
| 47 | 220 | 275 | 248 | 247 | 249 | 234 | |
| 48 | 210 | 231 | 229 | 227 | 231 | 205 | |
| 49 | 200 | 216 | 202 | 202 | 202 | 183 | |
| 50 | 150 | 82 | 77 | 74 | 80 | 73 | |
| 51 | 140 | 66 | 41 | 43 | 42 | 39 | |
Table 3: Calculated unscaled frequencies by HF/DFT (B3LYP&B3PW91) with 6-31+(d,p) and 6-311+G(d,p)basis sets.
Methyl group vibrations: The side chain of L-Valine has two methyl groups attached to the C atom. The CH3 stretching and deformation vibrations are more or less localized, and offer to good group frequencies. The positions of the C-H stretching vibrations are among the most stable in the spectrum. Since the CH3 group also exhibit CS symmetry. In aliphatic compounds, the asymmetric and symmetric CH3 stretching vibrations are normally observed in the region 2950- 2850 cm-1 [21-23]. In the present compound, the C-H stretching vibrations are found at 2920, 2910, 2890, 2880, 2860 and 2850 cm-1. The entire vibrational bands are situated in the lower part of the C-H vibrational region of the spectra. The first three of the above are asymmetric and rest of others are symmetric vibrations. The C-H in plane and out of plane bending vibrations is found normally in the region of 1250-1000 cm-1 and 970-700 cm-1 respectively [24,25]. Accordingly, the in plane bending group frequencies are located at 1190, 1170, 1165, 1140 and 1105 cm-1 and the out of plane bending sequences are identified at 820, 720, 710, 560, 500 and 450 cm-1. Except three of the out of plane bending, all the bending vibrations are observed within the expected region. This is because of the influence of amine group in the chain. The C-H vibrations are suppressed much since the present compound is amino acid.
Predicted by the DFT calculations, in aliphatic compounds containing CH3 group, the series of the bands appearing as asymmetric and symmetric deformation modes in the region 1400-1500 cm-1 [26-28] are mainly due to methyl deformation, coupling with the C-H and C-C stretching frequencies, two different extends and in different way. In the present study, the Raman bands at 1460 cm−1 (very strong) and 1450 cm−1 (strong) are attributed to the asymmetric deformation modes of isopropyl group. Appearance of these bands is due to presence of two independent CH3 groups in the amino acid residues in different environments. The symmetric deformation mode of isopropyl group normally exhibits relatively two very strong bands at 1400 and 1395 cm−1. They are due to the interaction between hydrogen atoms in two different methyl groups depending upon whether they are moving either closer or away from each other’s way [28]. The methyl twisting vibrational signals highlighted at 210 and 200 cm-1 in Raman spectrum.
C-CH3 Vibrations: The title molecule contains two methyl groups in molecular chain, there are two C-CH3 stretching vibrations are possible. The C-CH3 vibrations usually combine with C-H in-plane bending vibration. According to which, the active fundamentals are appeared with medium intensity at 940 cm-1 in IR and Raman are identified as C-CH3 stretching vibration. The C-CH3 in-plane bending vibration is found at 340 cm-1 and out-of-plane bending vibration is found at 260 cm-1. As reported in the literature [29,30], all the above C-CH3 vibrations deviated much from the expected range. This is purely due to the repulsion between methyl groups.
C-H vibrations: The C-H stretching mode of aldehyde group has its characteristic magnitude in the range 2900-2700 cm-1 [31,32]. In this case, the C-H stretching vibration is found at 2970 and 2930 cm-1 in IR and Raman spectra. The C-H in plane bending mode (CH rocking) and out of plane bending mode (CH wagging) are expected in the region 1420-1370 cm−1 and 1100-900 cm-1 for aldehyde and its derivatives [33-35]. In L-Valine, the in plane and out of plane bending vibrational modes are observed at 1350 and 1325 cm-1 and 870 and 850 cm-1. Except the stretching mode, the bending modes are found out the expected region which is due to the interaction of hydroxyl group at the nearest.
Amino group vibrations: One of the hydrogen bond of the amino group is removed and is attached with oxygen forms OH in the chain. Aliphatic primary amines salts are characterized by strong absorption in the region of 3200-2800 cm−1 due to the asymmetric and symmetric NH2+ stretching. Also the NH2+asymmetric and symmetric deformation wave numbers are expected to fall in the regions 1660-1610 cm−1 and 1550-1485 cm−1, respectively [36,37]. In this present case, the N-H stretching frequencies are observed at 3460 and 3350 cm−1. The first and second band is assigned to asymmetric and symmetric vibration. The NH2+asymmetric and symmetric deformation wave numbers are found at 1495 and 1465 cm-1. The out of plane bending vibrations are normally identified in the region 1150-900 cm−1 [38,39]. The out of plane bending vibrations are set up at 900 and 870 cm−1. Some of the amino group vibrations are concealed slightly due to the influence of carboxyl group. The NH2 twisting signal is raised at the last part of the spectrum.
COOH group vibrations: Free amino acids also have carboxilate ion (CO2- ion) stretching vibrations, a strong band occurring in the region 1600-1560 cm-1. Dicarboxylic acids have a strong band due to C=O stretching vibration of the carboxyl group at 1755-1700 cm-1 and another strong band at 1230-1215 cm-1 due to the stretching of the C-O bond [40,41]. According to the literature, two strong bands are identified at 1710 and 1270 cm-1 for C=O and C-O stretching vibrations respectively. C-O band is elevated up from the expected region due to the favoring of hydrogen bond.
The amino acid has hydroxyl stretching vibrations are generally [42] observed in the region around 3500 cm−1. The O-H group vibrations are likely to be the most sensitive to the environment, so they show pronounced shifts in the spectra of the hydrogen bonded species. The band due to the O–H stretching is of medium to strong intensity in the infrared spectrum, although it may be broad. The strong band appeared at 3550 cm−1 in the IR spectra is assigned to O–H stretching mode of vibration. The O–H in-plane bending vibration is observed in the region 1440–1260 cm−1 [43]. The O–H out-of-plane deformation vibration for phenol lies in the region 290-320 cm−1 for free O–H and in the region 517-710 cm−1 for associated O–H [38]. In both intermolecular and intra-molecular associations, the wavenumber is at higher value than that in the free O–H. The wavenumber increases with hydrogen bond strength because of large amount of energy required to twist the O–H bond [44]. The calculated values of in-plane/ out-of-plane bending vibrations of hydroxyl group are 1620 and 1375 cm−1, respectively. These modes are not observed experimentally. The carbonyl group is most important in the infrared spectrum because of its strong intensity of absorption and high sensitivity toward relatively minor changes in its environment. Intra- and intermolecular factors affect the carbonyl absorptions in common organic compounds due to inductive, mesomeric effects, field effects and conjugation effects. The COOH twisting vibration is found at 140 cm-1 concluded the Raman spectrum.
CCN vibrations: In amino acids, the absorption bands corresponding to C-C-N stretching vibrations are observed in the wave number region 1150-850 cm−1 [45,46]. In the title molecule, a strong Raman band at 935 cm−1 is attributed to the C-C-N symmetric stretching vibration. However the much-expected strong IR counterpart is not found distinctly as it overlaps with the degenerate stretching mode of the anion due to lifting of degeneracy. The strong peak at 1350 cm−1 is due to C-N antisymmetric stretching vibration. The wave numbers at 1050 and 900 cm−1 are attributed to C-C stretching vibrations. The in-phase and out-of-phase vibrations of skeletal carbon have also been identified 360 and 280 cm-1. These vibrations are degenerated with other bending vibrations.
NMR assessment
NMR spectroscopy is currently used for structure elucidation of complex molecules. The combined use of experimental and computational tools offers a powerful gadget to interpret and predict the structure of bulky molecules. The optimized structure of L-Valine is used to calculate the NMR spectra at B3LYP method with 6-311++G(d,p) level using the GIAO method and the chemical shifts of the compound are reported in ppm relative to TMS for 1H and 13C NMR spectra which are presented in Table 4. The corresponding spectra are shown in Figure 4.
| Atom position | B3LYP/6-311 +G(d,p) (ppm) | TMS B3LYP/ 6-311+G (2d,p) GIAO (ppm) | Shift (ppm) | B3LYP/6-311 +G(d,p) (ppm) | TMS B3LYP/ 6-311+G (2d,p) GIAO (ppm) | Shift (ppm) | B3LYP/ 6-311+ G(d,p) (ppm) | TMS B3LYP/ 6-311+G (2d,p) GIAO (ppm) | Shift (ppm) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Gas | DMSO | CCl4 | ||||||||
| C3 | 22.62 | 205.09 | 182.47 | 28.27 | 210.74 | 182.47 | 25.18 | 207.65 | 182.47 | |
| C4 | 120.66 | 61.80 | 58.86 | 118.88 | 63.57 | 55.31 | 119.71 | 62.74 | 56.97 | |
| C10 | 148.14 | 34.32 | 113.82 | 148.65 | 33.81 | 114.84 | 148.39 | 34.06 | 114.33 | |
| C12 | 163.67 | 18.79 | 144.88 | 164.76 | 17.70 | 147.06 | 164.19 | 18.26 | 145.93 | |
| C16 | 165.71 | 16.75 | 148.96 | 166.59 | 15.86 | 150.73 | 166.16 | 16.29 | 149.87 | |
| N6 | 211.70 | 46.69 | 165.01 | 211.68 | 46.71 | 164.97 | 211.56 | 46.83 | 164.73 | |
| O1 | 103.74 | 423.74 | 320 | 86.20 | 406.20 | 320 | 93.93 | 413.93 | 320 | |
| O2 | 248.16 | 568.16 | 320 | 135.97 | 455.97 | 320 | 194.73 | 514.73 | 320 | |
| H5 | 28.85 | 3.03 | 25.82 | 28.82 | 3.05 | 25.77 | 28.87 | 3.00 | 25.87 | |
| H7 | 28.16 | 3.71 | 24.45 | 27.60 | 4.27 | 23.33 | 27.96 | 3.91 | 24.05 | |
| H8 | 28.17 | 3.70 | 24.47 | 27.97 | 3.90 | 24.07 | 28.09 | 3.78 | 24.31 | |
| H9 | 28.69 | 3.18 | 25.51 | 28.03 | 3.84 | 24.19 | 28.35 | 3.52 | 24.83 | |
| H11 | 31.15 | 0.72 | 30.43 | 30.99 | 0.88 | 30.11 | 31.09 | 0.78 | 30.31 | |
| H13 | 29.92 | 1.95 | 27.97 | 30.76 | 1.12 | 29.64 | 30.26 | 1.61 | 28.65 | |
| H14 | 31.74 | 0.13 | 31.61 | 31.55 | 0.32 | 31.23 | 31.66 | 0.21 | 31.45 | |
| H15 | 31.74 | 0.14 | 31.6 | 31.80 | 0.07 | 31.73 | 31.76 | 0.11 | 31.65 | |
| H17 | 32.14 | 0.26 | 31.88 | 31.59 | 0.28 | 31.31 | 31.93 | 0.05 | 31.88 | |
| H18 | 31.96 | 0.08 | 31.88 | 31.87 | 0.01 | 31.86 | 31.93 | 0.05 | 31.88 | |
| H19 | 31.13 | 0.74 | 30.39 | 31.20 | 0.68 | 30.52 | 31.14 | 0.73 | 30.41 | |
Table 4: Calculated 1H and 13C NMR chemical shifts (ppm) of L-Valine.

Figure 4: Experimental and calculated 1H and 13C NMR of L-Valine.
In view of the range of 13C NMR chemical shifts for similar organic molecules usually is >100 ppm [47,48] the accuracy ensures reliable interpretation of spectroscopic parameters. In the present work, 13C NMR chemical shifts of some carbons in the chain are >100 ppm, as they would be expected (Table 5).
| Atom position | B3LYP/6-311+G(d,p) (ppm) | TMS B3LYP/6-311+G(2d,p) GIAO (ppm) | Shift (ppm) | Experimental value |
|---|---|---|---|---|
| C3 | 22.62 | 205.09 | 182.47 | 175.0 |
| C4 | 120.66 | 61.80 | 58.86 | 30.0 |
| C10 | 148.14 | 34.32 | 113.82 | 125.0 |
| C12 | 163.67 | 18.79 | 144.88 | 150.0 |
| C16 | 165.71 | 16.75 | 148.96 | 150.0 |
| N6 | 211.70 | 46.69 | 165.01 | 175.0 |
| O1 | 103.74 | 423.74 | 320 | - |
| O2 | 248.16 | 568.16 | 320 | - |
| H5 | 28.85 | 3.03 | 25.82 | 19.0 |
| H7 | 28.16 | 3.71 | 24.45 | 19.0 |
| H8 | 28.17 | 3.70 | 24.47 | 19.0 |
| H9 | 28.69 | 3.18 | 25.51 | 19.0 |
| H11 | 31.15 | 0.72 | 30.43 | - |
| H13 | 29.92 | 1.95 | 27.97 | - |
| H14 | 31.74 | 0.13 | 31.61 | - |
| H15 | 31.74 | 0.14 | 31.6 | - |
| H17 | 32.14 | 0.26 | 31.88 | - |
| H18 | 31.96 | 0.08 | 31.88 | - |
| H19 | 31.13 | 0.74 | 30.39 | - |
Table 5: Experimental and Calculated 1H and 13C NMR of L-Valine.
In the case of L-Valine, the chemical shift of C3, C4, C10, C12 and C16 are 182.47, 58.86, 113.82, 144.88 and 148.96 ppm respectively. The shift is less in C4 (expt. 30 ppm) than rest of others. This is mainly due to the breaking of paramagnetic shield of proton by the substitutions of CH3 and COOH. The C3 in the chain has more shifted than other due to the delocalization of σ and π electrons. The shift of the entire carbons of the ring is found increased when going from gas to solvent due to the solvent effect. The shift values of carbons in DMSO are greater than CCl4 solvent. The chemical shift values of oxygen have not changed due to the solvent effect. The experimental and theoretical 1H and 13C NMR chemical shift of L-Valine are presented in Table 5. From the Table 5, it is clear that, the experimental values of chemical shift are slightly more than calculated values. The chemical shift is greater in C16 than C12 since the attraction of amino group.
This effect of isolation is the main cause to change the chemical property from amino acid to L-Valine. There is no change of chemical shift in N and O due to the rigidity of the diamagnetic shielding of the atom. From the observation, it is clear that the change of chemical property of L-Valine is only in favor of CH3 groups. In addition to that, due to the accessibility of CH3 groups, the amino acid itself is disrupted. This view is also evident that the entire property of the amino acid is deflected towards L-Valine.
Electronic properties (frontier molecular analysis)
The frontier molecular orbitals are very much useful for studying the electric and optical properties of the organic molecules. The stabilization of the bonding molecular orbital and destabilization of the antibonding can increase when the overlap of two orbitals increases. In the molecular interaction, there are the two important orbitals that interact with each other. One is the highest energy occupied molecular orbital is called HOMO represents the ability to donate an electron. The other one is the lowest energy unoccupied molecular orbital is called LUMO as an electron acceptor. These orbitals are sometimes called the frontier orbitals. The interaction between them is much stable and is called filled empty interaction.
The 3D plots of the frontier orbitals, HOMO and LUMO for L-Valine molecule are in gas, shown in Figures 5 and 6. According to Figure 5, the HOMO is mainly localized over the Nitrogen, carbons of amino and a methyl group which connects two NH2 and CH3 groups in the chain. The entire C in the chain is connected by SP3 orbital lobes. The SP3 orbital lobe of C overlapped with SP3 of O of nearby COOH group. However, LUMO is characterized by a charge distribution connects the entire atoms of CH3, NH2 and COH in the same plane as an umbrella. When the two same sign orbitals overlap to form a molecular orbital, the electron density will occupy at the region between two nuclei. The molecular orbital resulting from in-phase interaction is defined as the bonding orbital which has lower energy than the original atomic orbital. The out of phase interaction forms the anti bonding molecular orbital with the higher energy than the initial atomic orbital. From this observation it is clear that the in and out of phase interaction are present in HOMO and LUMO respectively. The HOMO→LUMO transition implies an electron density transferred among CH3, NH2 and COOH groups. The HOMO and LUMO energy are 7.265 eV and 0.1670 eV in gas phase (Figure 5). Energy difference between HOMO and LUMO orbital is called as energy gap (kubo gap) that is an important stability for structures. The DFT level calculated energy gap is 7.098 eV, show the large energy gap and reflect the zero electrical activity of the molecule.

Figure 5: Frontier molecular orbitals, Homo and Lumo for L-Valine.

Figure 6: Frontier molecular orbitals of L-Valine: Humo and Lumo in UV region.
Optical properties (HOMO-LUMO analysis)
The UV and visible spectroscopy is used to detect the presence of chromophores in the molecule and whether the compound has NLO properties or not. The calculations of the electronic structure of L-Valine are optimized in singlet state. The low energy electronic excited states of the molecule are calculated at the B3LYP/6-311++G(d,p) level using the TD-DFT approach on the previously optimized ground-state geometry of the molecule. The calculations are performed for L-Valine in gas phase and with the solvent of ethanol, methanol and DMSO. The calculated excitation energies, oscillator strength (f) and wavelength (λ) and spectral assignments are given in Table 6. The major contributions of the transitions are designated with the aid of SWizard program [49].
| λ (nm) | E (eV) | ( f ) | Major contribution | Assignment | Region | Bands |
|---|---|---|---|---|---|---|
| Gas | ||||||
| 434.66 | 2.852 | 0.001 | H→L (92) | n→π* | Visible | R-band (German, radikalartig) |
| 314.21 | 3.946 | 0.002 | H→L (89) | n→π* | Quartz UV | |
| 312.12 | 3.962 | 0.002 | H→L (86) | n→π* | Quartz UV | |
| DMSO | ||||||
| 271.82 | 4.561 | 0.003 | H→L-1 (90) | n→π* | Quartz UV | R-band (German, radikalartig) |
| 230.01 | 5.390 | 0.009 | H→L-1 (90) | n→π* | Quartz UV | |
| 228.07 | 5.436 | 0.002 | H→L-1 (87) | n→π* | Quartz UV | |
| 204.60 | 6.059 | 0.063 | H+1→L-1 (83) | σ→σ* | Quartz UV | |
| Methanol | ||||||
| 272.39 | 4.551 | 0.0003 | H→L-1 (86) | n→π* | Quartz UV | R-band (German, radikalartig) |
| 231.30 | 5.360 | 0.009 | H→L-1 (85) | n→π* | Quartz UV | |
| 228.26 | 5.431 | 0.002 | H→L-1 (78) | n→π* | Quartz UV | |
| 207.42 | 5.977 | 0.068 | H+1→L-1(77) | σ→σ* | Quartz UV | |
| Ethanol | ||||||
| 273.0 | 4.541 | 0.0003 | H+1→L (86) | n→π* | Quartz UV | R-band (German, radikalartig) |
| 232.72 | 5.327 | 0.009 | H+1→L-1 (85) | n→π* | Quartz UV | |
| 228.50 | 5.425 | 0.002 | H→L-1 (78) | n→π* | Quartz UV | |
| 207.89 | 5.964 | 0.069 | H+1→L-1(74) | σ→σ* | Quartz UV | |
| Acetone | ||||||
| 273.54 | 4.532 | 0.0004 | H+1→L (90) | n→π* | Quartz UV | R-band (German, radikalartig) |
| 234.01 | 5.298 | 0.009 | H→L-1 (83) | n→π* | Quartz UV | |
| 228.71 | 5.421 | 0.002 | H→L-1 (88) | n→π* | Quartz UV | |
| 208.22 | 5.954 | 0.067 | H+1→L-1(86) | σ→σ* | Quartz UV | |
H: HOMO; L: LUMO
Table 6: Theoretical electronic absorption spectra of L-Valine (absorption wavelength λ (nm), excitation energies E (eV) and oscillator strengths (f) using TD-DFT/B3LYP/6-311++G(d,p) method.
TD-DFT calculations predict three transitions in the near Visible and quartz ultraviolet region. In the case of gas phase, the strong transition is at 434.66, 314.21 and 312.12 nm with an oscillator strength f=0.01, 0.002 with 3.946 eV energy gap. The transition is n → π* in visible and quartz ultraviolet region. The designation of the band is R-band (German, radikalartig) which is attributed to above said transition of single chromophoric groups, such as carbonyl group. They are characterizes by low molar absorptivities (ξmax<100) and undergo hypsochromic shift with an increase in solvent polarity. The simulated UV-Visible spectra in gas and solvent phase of L-Valine are shown in Figure 7.

Figure 7: UV-Visible Spectra of L-Valine in gas and solvent phase.
In the case of DMSO solvent, strong transitions are271.82, 230.01, 228.07 and 204.60 nm with an oscillator strength f=0.003, 0.009, 0.002 and 0.63 with maximum energy gap 6.059 eV. They are assigned to n → π* and σ→ σ* transitions and belongs to quartz ultraviolet region. This shows that, from gas to solvent, the transitions moved from visible to quartz ultraviolet region. This view indicates that, the L-Valine molecule colored and it is capable of having rich NLO properties. In addition to that, the calculated optical band gap 3.45 eV which ensure that the present compound has NLO properties. In view of calculated absorption spectra, the maximum absorption wavelength corresponds to the electronic transition from the HOMO to LUMO with maximum contribution. In this present compound, the chromophores is CH3 group, the properties are changed and enhanced from free amino acid to L-Valine by adding CH3 group further.
The chemical hardness and potential, electronegativity and Electrophilicity index are calculated and their values are shown in Table 7. The chemical hardness is a good indicator of the chemical stability. The chemical hardness is decreased slightly (1.72-3.01) in going from Gas to solvent. Hence, the present compound has much chemical stability. Similarly, the electronegativity is increased from 3.45 up to 3.36, from Gas to solvent, if the value is greater than 1.7; the property of bond is changed from covalent to ionic. Accordingly, the bonds in the compound converted from covalent to ionic and are independent of solvent. Electrophilicity index is a measure of energy lowering due to maximal electron flow between donor [HOMO] and acceptor [LUMO]. From the Table 7, it is found that the Electrophilicity index of L-Valine is 3.45 in gas and 3.36 in solvent, which is moderate and this value ensure that the strong energy transformation between HOMO and LUMO. The dipole moment in a molecule is another important electronic property. Whenever the molecule has larger the dipole moment, the intermolecular interactions are very strong. The calculated dipole moment value for the title compound is 12.24 Debye in gas and 15.75 in solvent. It is too high which shows that; the L-Valine molecule has strong intermolecular interactions.
| TD-DFT/B3LYP/ 6-311G++(d,p) | Gas | DMSO | Ethanol | Methanol |
|---|---|---|---|---|
| Etotal (Hartree) | -402.41 | -402.47 | -402.47 | -402.47 |
| EHOMO (eV) | 5.1810 | 6.3797 | 6.3590 | 6.3383 |
| ELUMO (eV) | 1.7279 | 0.3559 | 0.3673 | 0.3804 |
| ΔEHOMO-LUMO gap (eV) | 3.453 | 6.023 | 5.991 | 5.9597 |
| EHOMO-1 (eV) | 5.627 | 6.666 | 6.777 | 6.7574 |
| ELUMO+1 (eV) | 0.3439 | 0.0816 | 0.0772 | 0.0723 |
| ΔEHOMO-1-LUMO+1 gap (eV) | 5.284 | 6.584 | 6.699 | 6.684 |
| Chemical hardness (h) | 1.7265 | 3.0119 | 2.9958 | 2.9789 |
| Electronegativity (χ) | 3.4544 | 3.3678 | 3.36315 | 3.3593 |
| Chemical potential (μ) | 0.514 | 0.617 | 0.614 | 0.611 |
| Chemical softness (S) | 0.2895 | 0.1660 | 0.16690 | 0.1678 |
| Electrophilicity index (ω) | 3.455 | 1.882 | 1.859 | 1.894 |
| Dipole moment | 12.245 | 15.759 | 15.703 | 15.646 |
Table 7: Calculated energies values, chemical hardness, electro negativity, Chemical potential, Electrophilicity index of L-Valine from UV-Visible region.
Molecular electrostatic potential (MEP) maps
The molecular electrical potential surfaces illustrate the charge distributions of molecules three dimensionally. This map allows us to visualize variably charged regions of a molecule. Knowledge of the charge distributions can be used to determine how molecules interact with one another and it is also be used to determine the nature of the chemical bond. Molecular electrostatic potential is calculated at the B3LYP/6-311+G(d,p) optimized geometry. There is a great deal of intermediary potential energy, the non red or blue regions indicate that the electro negativity difference is not very great. In a molecule with a great electro negativity difference, charge is very polarized, and there are significant differences in electron density in different regions of the molecule. This great electro negativity difference leads to regions that are almost entirely red and almost entirely blue. Greater regions of intermediary potential, yellow and green, and smaller or no regions of extreme potential, red and blue, are key indicators of a smaller electronegativity.
The color code of these maps is in the range between -6.15 a.u. (deepest red) to 6.15 a.u. (deepest blue) in compound. The positive (blue) regions of MEP are related to electrophilic reactivity and the negative (green) regions to nucleophilic reactivity shown in Figure 8. As can be seen from the MEP map of the title molecule, the negative regions are mainly localized on single and double oxygen atoms. A maximum positive region is localized on the methyl groups indicating a possible site for nucleophilic attack. The MEP map shows that the negative potential sites are on electronegative atoms (O atom) as well as the positive potential sites are around the methyl groups. From these results, it is clear that the methyl groups indicate the strongest attraction and carboxylic group indicates the strongest repulsion.

Figure 8: Molecular electrostatic potential and contour map of L-Valine.
Polarizability and First order hyperpolarizability calculations
In order to investigate the relationships among molecular structures and non-linear optic properties (NLO), the polarizabilities and first order hyperpolarizabilities of the L-Valine compound was calculated using DFT-B3LYP method and 6-311+G(d,p) basis set, based on the finite-field approach.
The polarizability and hyperpolarizability tensors (αxx, αxy, αyy, αxz, αyz, αzz and βxxx, βxxy, βxyy, βyyy, βxxz, βxyz, βyyz, βxzz, βyzz, βzzz) can be obtained by a frequency job output file of Gaussian. However, α and β values of Gaussian output are in atomic units (a.u.) so they have been converted into electronic units (esu) (α; 1 a.u.=0.1482×10−24 esu, β; 1 a.u.=8.6393×10−33 esu). The mean polarizability (α), anisotropy of polarizability (Δα) and the average value of the first hyperpolarizability 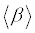 can be calculated using the equations.
can be calculated using the equations.
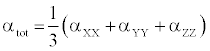
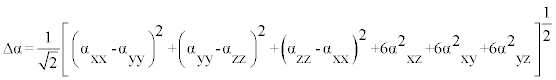
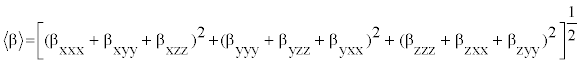
In Table 8, the calculated parameters described above and electronic dipole moment {μi (i=x, y, z) and total dipole moment tot μ } for title compound are listed. The total dipole moment is be calculated using the following equation
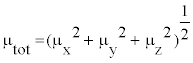
| Parameter | a.u. | Parameter | a.u. |
|---|---|---|---|
| αxx | 58.515 | βxxx | 8.575 |
| αxy | 3.810 | βxxy | -6.711 |
| αyy | 51.13 | βxyy | 3.304 |
| αxz | 1.11 | βyyy | -20.014 |
| αyz | 0.899 | βxxz | 0.717 |
| αzz | 47.92 | βxyz | -2.743 |
| αtot | 82.60 | βyyz | 0.216 |
| Δα | 102.05 | βxzz | -4.166 |
| μx | 3.669 | βyzz | -3.960 |
| μy | -4.021 | βzzz | 0.0395 |
| μz | 0.5156 | βtot | 97.08 |
| μ | 5.468 |
Table 8: The dipole moments μ (D), the polarizability α (a.u.), the average polarizability αo (esu), the anisotropy of the polarizability Δα (esu), and the first hyperpolarizability β (esu) of L-Valine.
It is well known that, molecule with high values of dipole moment, molecular polarizability, and first hyperpolarizability having more active NLO properties. The first hyperpolarizability (β) and the component of hyperpolarizability βx, βy and βz of L-Valine along with related properties (μ0, αtotal, and Δα) are reported in Table 8. The calculated value of dipole moment is found to be 5.468 Debye. The highest value of dipole moment is observed for component μX. In this direction, this value is equal to 3.66 D. The lowest value of the dipole moment of the molecule compound is μY component (-4.02 D). The calculated average polarizability and anisotropy of the polarizability is 82.60×10-24 esu and 102.05×10−24 esu, respectively. The magnitude of the molecular hyperpolarizability β, is one of important key factors in a NLO system. The B3LYP/6-311+G(d,p) calculated first hyperpolarizability value (β) is 97.08×10−30 esu. From the above results, it is observed that, the molecular Polarizability and hyperpolarizability of the title compound in all coordinates are active. So that, the L-Valine can be used to prepare NLO crystals and those crystal is able to produce second order harmonic waves with more amplitude.
In the present investigation, FT-IR, FT-Raman and 13C NMR and 1H NMR spectra of the L-Valine are recorded and the observed vibrational frequencies are assigned depending upon their expected region. The chronological change of finger print and group frequency region of the amino acid with respect to the functional group has also monitored. The change of geometrical parameters along with the substitutions is deeply analyzed. The simulated 13C NMR and 1H NMR are compared with the recorded spectrum and the chemical shifts related to TMS are studied. The electrical and optical properties of the L-Valine are profoundly investigated using frontier molecular orbital. From the UV-Visible spectra, it is found that the present compound is optically active and posses NLO properties. The molecular electrostatic potential (MEP) map is performed and from which the change of the chemical properties of the compound is also discussed.