Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2010) Volume 3, Issue 5
Arti fi cial Neural Networks (ANNs) are simpli fi ed models of the nervous system, in which neurons are considered as simple processing units linked with weighted connections called synaptic ef fi cacies. These weights are gradually adjusted according to a learning algorithm.Oxidoreductase any of a class of enzymes that catalyse oxidation–reduction reactions, i.e. they are involved in the transfer of hydrogen or electrons between molecules. They include the oxidases and dehydrogenases.
In this paper, an attempt has been made to develop a neural network-based method for predicting the secondary structure of protein ( Human Oxidoreductase family). The neural network has been trained using Bayesian Regularization Feed-forward Backpropagation Neural Network Technique to predict the -helix, -sheet and coil regions of this protein family. Feed-forward neural network have been trained by analyzing windows of 25 parameters for predicting the central residue of protein sequence. PSI-BLAST has been used for multiple-sequence alignment. SCOP and PDB database has been used for searching the primary and secondary structure of proteins and for training the data set. The method correctly identi fi es the secondary structure of Human Oxidoreductase family with more than 79% accuracy, which is well above any previously reported method.
Keywords: Neural network; Human oxidoreductase; Protein; Secondary structure
The most important level of protein structure is the secondary structure which is mainly composed of alpha helices, beta strands and coils, which are formed from local sequences of amino acids (Branden and John, 1991). Knowing a protein’s secondary structure helps to determine the structural properties of that protein. Several methods have been developed to determine secondary structure, with varying accuracy. One method involves analyzing the X-ray diffraction patterns of crystallized proteins. While X-ray diffraction is rather time-consuming, it is extremely accurate (Qian and Sejnowski, 1988). Another method, structure homology, or threading, utilizes an amino acid sequence with a known secondary structure as a model to predict the secondary structure of another similar sequence (Holley and Karplus, 1989). Various theoretical algorithms with high accuracy have also been proposed. Two of the most prominent are the DSSP and Chou-Fasman algorithms. The first algorithm determines secondary structure through knowledge obtained from the three dimensional protein structure, such as hydrogen bonds and various geometrical features (Kabsch and Sander, 1983). On the other hand, the Chou-Fasman algorithm predicts secondary structure by using many empirically determined rules in addition to information concerning the primary sequence (Chou and Fasman, 1974).
A more recent and interesting approach to secondary structure prediction has been the use of neural networks, which have been found to have respectable accuracy (Qian and Sejnowski, 1988). These information-processing systems consist of a large number of simple interconnected processing units that operate in parallel. All of these units are found in three different types of layers in the network: the input layer, the output layer, and in some cases, the hidden layers in between the input and output layers (Laurence, 1994). Each unit has an internal activation state which fluctuates according to the unit’s input: excitatory input increases the activation, while inhibitory input decreases the activation (Khanna, 1990). By changing the activation of the units, neural networks are capable of learning by assimilating past inputs into the activation of each unit (Rumelhart and McClelland, 1986). This capability has led to numerous applications in areas such as signal processing and pattern and speech recognition (Laurence, 1994). This study was aimed to develop an improved fully-automated method for the prediction of physiochemical properties of catalytic residues of structural protein of PDB using a carefully selected and supervised Machine learning Backpropagation algorithm coupled with an optimal discriminative set of structural protein properties. This study helps in denovo prediction of properties of functional sites of proteins (Yadav et al., 2009). The prediction of ß-turns is an important element of protein secondary structure prediction. Recently, a highly accurate neural network based method Betatpred2 has been developed for predicting β-turns in proteins using positionspecific scoring matrices (PSSM) generated by PSI-BLAST and secondary structure information predicted by PSIPRED (Harpreet and Raghava, 2004). In this paper we evaluate the effects of an imbalanced data set in training and learning of neural networks when they are applied to predict protein secondary structure. For this we applied resampling methods to tackle the imbalance class problem. Results show that imbalanced data sets decrease the helixes predictions rates. Although, protein data set distribution does not affect significantly the global accuracy (Q3) (Palodeto et al., 2009).
An artificial neural network (ANN) solution is described for the recognition of domains in protein sequences. A querysequence is first compared to a reference database of domain sequences byuse of BLAST and the output data, encoded in the form of six parameters, are forwarded to feed-forward artificial neural networks with six input and six hidden units with sigmoidal transfer function. The recognition is based on the distribution of BLAST scores precomputed for the known domain groups in a database versus database comparison (Murvai et al., 2001).
In this paper, an attempt has been made to improve the predictive capabilities of neural networks for protein secondary structure with the use of predictions made by the DSSP and Chou- Fasman algorithms. With this intent, we evaluate the accuracy that the network achieves while using information obtained from either the DSSP or Chou-Fasman algorithms, in determining whether or not the secondary structure prediction of a given amino acid sequence is valid. The architecture of the network, which was constructed using the Bayesian Regularization Backpropagation Function of MATLAB 7.0, is described and the results produced by the network are discussed and statistically analyzed (Figure 1).

Figure 1
Network design
The neural network that was used in this investigation consists of a twenty five-unit input layer and three-unit output layer. No hidden layers were incorporated into the network due to the conclusion of Qian and Sejnowski (1988) that the peak performance of their network in determining protein secondary structure was nearly independent of the number of hidden units. Furthermore, the network utilizes a feed-forward design, in which signals are transferred forward from the input units to the output unit (Kneller et al., 1990).
The twenty five units in the input layer encode a window of twenty five residues of an amino acid sequence, composed of twelve residues on either side of the central residue. The output unit represents the prediction made by the neural network as to whether the central residue represents alpha helix, beta sheet or coil.
The activation state of each unit, Xi, is a real value between 0 and 1. The strength of the connection, or weight, between a unit j and another unit i is represented by a real number Wij . The activation of a unit can be calculated by summing the products of every unit’s output Yj and weight Wij and then adding a bias term, bj :
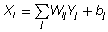
Having calculated the activation Xi for a unit, the output of that unit, Yi can be computed using the logistic sigmoid function
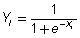
and then propagated to the next layer of the neural network.
During each cycle, the inputs are presented to the network. The weights of the units are adjusted at the end of the cycle, and this procedure is repeated. Back-propagation, a type of learning algorithm, is used to optimize the adjustment of the weights. This form of supervised training, in which the desired output is presented to the network along with the inputs (Laurence, 1994), was used to train the neural network as shown in Figure 2.

Figure 2: Bayesian Regularization Feed-forward Backpropagation Neural Network showing the connections between the computing units
Network training and testing sets
To train and test the neural network, the amino acid sequences of Human Oxidoreductase protein, were obtained from the Protein Data Bank (PDB) at Brookhaven National Laboratory (Bernstein et al., 1977) as shown in Table 1.
| PDB ID | Length | Classification |
|---|---|---|
| 1grh | 141 | Low resolution protein structure |
| 1alg | 24 | Peptide |
| 1hfp | 185 | Alpha and Beta protein |
| 1hfq | 186 | Alpha and Beta protein |
| 1hfr | 186 | Alpha and Beta protein |
| 1ohk | 186 | Alpha and Beta protein |
| 1auc | 105 | Alpha and Beta protein |
| 1dsw | 152 | All Beta |
| 1mar | 315 | Alpha and Beta protein |
| 1kmg | 153 | All Beta |
Table 1: List of PDB ID, their length of sequence and classification based on SCOP database for Human Oxidoreductase family proteins
The creation of input patterns for propagation through the network was accomplished by MATLAB program. The program first parses the secondary structure predictions and identifies the residues that have different predictions, of which one of the following: alpha helix, beta sheet, or coil. Next, it takes each identified residue, the twelve residues on either side of it and converts those twelve values using a numerical encoding scheme into a format understandable by the network. Table 2 show the schemes that were used to convert the amino acids into numerical formats.
| Input | A | C | D | E | ... | T | V | W | Y |
|---|---|---|---|---|---|---|---|---|---|
| Output | 0.00 | 0.05 | 0.10 | 0.15 | ... | 0.85 | 0.90 | 0.95 | 1.00 |
Table 2: Amino Acid Encoding Scheme
Table 3 show the schemes that were used to convert the secondary structure predictions into numerical formats.
| DSSP Classes | Predicted Class | Numerical Encoding |
|---|---|---|
| H,G | helix | 0.33 0.66 1.00 |
| E | Strand | 1.00 0.66 0.33 |
| B, T, S, I, e, g, h | coil | 0.66 1.00 0.33 |
Table 3: Secondary Structure Encoding Scheme.
At this point, we have a comprehensive set of input patterns for a particular protein. Finally, the program creates input patterns by selecting an element for each of the twenty five inputs from all of the elements with the same input positions in the input patterns that have just been determined.
The training and testing sets were compiled on Human Transferase family proteins.
During each of the tests, the neural network was trained for maximum of one hundred ephocs using the appropriate training set before the predictions were made for the testing set. A typical training set contains over n windows per protein chains comprising of more than n x l training patterns in total, where ‘l’ is the length of the sequence. A typical architecture is a fully-connected network (25 inputs, 3 outputs). The prediction is determined by the strongest of three network outputs. For example, the output (-1.83e-15, -1.07e- 16, 1) is taken to be a Coil prediction. The results of the tests are shown in Table 4.
| Window No. | Estimated SS from PDB | Observed SS from NN |
|---|---|---|
| 1 | C | C |
| 2 | C | C |
| 3 | E | C |
| 4 | C | C |
| 5 | E | E |
| 6 | E | E |
| 7 | C | C |
| 8 | E | E |
| 9 | C | C |
| 10 | C | C |
| 11 | E | E |
| 12 | C | E |
| 13 | C | C |
Table 4: Performance results of neural network using unknown protein sequence
To measure the performance of neural network, the correlation coefficient for each target class has been calculated as follows:
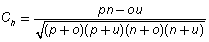
where, Ch = Correlation coefficient for helix
p = patterns correctly assigned to helix
n = patterns correctly assigned to non –helix
o = patterns incorrectly assigned to helix
u = patterns incorrectly assigned to not – helix
The correlation coefficients for helix (Ch) was found to be 0.54 for strand (Ce) it was 0.83 and for coil (Cc) it was 0.42.
Perfect prediction of protein secondary structures is probably impossible for a variety of reasons including the fact that a conformation may also depend on other environmental variables, related to solvent, acidity, hydrophobicity, hydrophilicity and so forth. It is however comforting to observe that steady progress is being made in this area, with an increasing number of secondary structures being predicted in the structural databases, and steady improvement of classification and machine learning methods. Here, neural network architecture has been developed that predicts secondary structure of protein with a performance of almost 79% correct prediction. This neural network require a smaller training time compared to fully connected networks with the same number of units. The analysis of the results have demonstrated that the development of a better multi-expert system architecture with different representation schemes can yield a better and more promising solution.
Work is also under way to improve the sequence-to-structure prediction produced by the neural network and can be applied to other families of protein.