Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2011) Volume 4, Issue 2
Sub-cellular localization of proteins is crucial for the dynamic life of cells. Its ascertainment is an important step to elucidate proteins' biological functions. Various experimental and computational methods have been developed for this purpose. Using a Bayesian model, we integrated five sub-modules based on different protein features, such as homology, amino acid composition, sorting signals and functional motifs, to predict sub-cellular localization of non-plant eukaryotic protein. This method has higher accuracy and Matthew's correlation coefficient values than previous algorithms against five independent test datasets, and is able to predict efficiently nine major sub-cellular compartments for both single-localized and multiple-localized proteins. As an application, we also combined this method with the proteome mass-spectrum quantitative information, improving the performance of PreLocABC dramatically. This method has been developed into an online prediction system (PreLocABC). Users may submit their protein sequences online, and the prediction results for protein sub-cellular localization will be returned. The web interface of PreLocABC is available at http://61.50.138.123/PreLocABC.
Keywords: Bayesian model; Sub-cellular localization; Classifier; Bioinformatics
MCC: Matthew's Correlation Coefficient; KNN: K-Nearest Neighbors; GFP: Green Fluorescent Protein; PCP: Protein Correlation Profiling; SP: SWISS-PROT; mTP: mitochondrial Targeting Peptides; cTP: chloroplast Transit Peptides
Eukaryotic proteins are organized into sub-cellular compartments that generate appropriate environments for their specialized functions. Thus, assigning certain localization to proteins in cells is an important step to elucidate their biological function, especial for those uncharacterized proteins.
Over the last few years, there have been numerous experimental approaches attempting to determine protein sub-cellular localization. Fusion to Green Fluorescent Protein (GFP) has been used to localize proteins through fluorescence screening of transfected yeast cells or mammalian cells. However, the GFP can interfere with the proper localization of proteins directed by sequence or structural signals [1]. The development of mass spectrometry-based proteomics technology resulted in another experimental method. Cells or organs are firstly homogenized and fractionated, and the composition proteins are identified by mass spectrometry. Although it is able to identify the most abundant proteins of a particular organelle with proper centrifugation and fractionation techniques [2-4], this approach is susceptible to cross-compartment contamination. To solve this problem, Andersen et al. [2] introduced a method named Protein Correlation Profiling (PCP) which utilizes protein quantitative data from mass spectrum as the characteristic of their sub-cellular location, thus reducing the need for a complete purification of each fraction [5]. Together with the experimental approaches, computational methods offer a complementary and comprehensive approach. The existing computational methods usually combine several of the following features or information to produce prediction results:
(1) Signal sequences, which direct the proteins to their proper subcellular localization. These signal sequences include signal peptides, mitochondrial Targeting Peptides (mTP), chloroplast Transit Peptides (cTP) and nuclear import signals. Existing algorithms mainly based on signal sequences such as SignalP [6], MitoProt [7], TargetP [8-9], Nucleo [10], PSORT [11] and Predotar (www.inra.fr/predotar/) identify signal sequences specific to the endoplasmic reticulum (ER), mitochondria/chloroplast and in some cases predict proteins that target to multiple compartments.
(2) Gglobal/local sequence features, such as amino acid composition (MITOPRED [12]) and functional domains (pTARGET [13]), which differ between proteins in different sub-cellular compartments.
(3) Homology, including methods using phylogenetic profiling of proteins [17], which can be applied to the prediction of proteins in organelles with endo-symbiotic origins and to those with common protein motifs. Proteome Analyst [18] is a recent addition to this category, which uses a naïve Bayesian network to predict localization mostly based on the SWISS-PROT keywords and annotations that can be extracted from the closest homolog of query proteins.
(4) External information, such as GO and SP annotations [14] and protein-protein interaction data [15,16].
Details of the characteristics of the existing predictors for protein sub-cellular localization have been summarized in Supporting materials\supplement.doc\Table S1.
Here we introduce PreLocABC, a novel predictor of protein subcellular localization using a Bayesian classifier for non-plant eukaryotic proteins, which integrates five sub-modules by a Bayesian model to convert the amino acid sequences of a protein into different localization features, such as homology, amino acid composition, sorting signals and functional motifs. This method has better performance than previous algorithms against five independent test datasets, and is able to predict efficiently nine major sub-cellular compartments for both singlelocalized and multiple-localized proteins. Furthermore, PreLocABC is not only a predictor of protein sub-cellular localization, but also an integration strategy. In addition to integrating the five sub-modules mentioned above, it can also integrate other different localization features as needed. For example, the PCP-KNN sub-module was constructed in the present study to integrate the high-throughput mass spectrum quantitative information of proteins into PreLocABC to improve its prediction efficiency.
Datasets
(1) The human and mouse protein sequence datasets were downloaded from SWISS-PROT (SP) [19] database, release 2010_04 (http://www.ebi.ac.uk/swissprot). These proteins were assigned to certain sub-cellular localization in the nine major compartments (cytoplasm, ER, extracellular space, Golgi apparatus, lysosome, mitochondrion, nucleus, peroxisome and plasma membrane) based on the annotation in the comments (CC) section. We chose protein sequences from the human species (SP_human dataset) and mouse species (SP_mouse dataset) as the training dataset and one of the independent test datasets of PreLocABC, respectively. In order to obtain a high-quality training dataset and independent test dataset, we filtered the data as follows (i) We removed sequences with ambiguous and uncertain annotations such as 'by similarity', 'potential', 'probable' and 'possible'. (ii) We clustered sequences at 80% identity using the cdhit program [20] and removed highly homologous sequences from SP_ human dataset in order to get rid of the homologues and redundancy bias [13,21]. (iii) For all sequences in SP_mouse dataset, we run BLASTP in an all-on-all fashion to remove the redundancy between training and testing datasets. We used NCBI BLAST (Version 2.2.8, ftp://ftp.ncbi.nlm.nih.gov/blast/), the identity of blast was set to 85%, the e-value was set to 10.0, and the Matrix used was blosum62. Finally, we extracted a total of 5646 human and 5366 mouse protein sequences.
(2) The Hera dataset was obtained from Human ER Apercu (Hera) and the supporting materials of M Scott, et al. [22] (http://www.mcb. mcgill.ca/∼hera), which is a comprehensive database that contains 2214 human organelle proteins annotated with sub-cellular localization information with a high degree of certainty (classification criteria "c" or "e" in Hera). For all sequences in this database, we also removed the redundancy between training and Hera datasets by NCBI BLAST. After that, a total of 1124 human protein sequences (Hera_1124 dataset) were extracted and used as one of the independent test datasets for PreLocABC.
(3) LOCATE datasets were obtained by extracting human and mouse protein sequences from LOCATE [23] (http://locate.imb.uq.edu. au, release on Sept. 20, 2007). We selected proteins from the LOCATE database, which is annotated with literature-mined localization data, because these sets are determined by manual review and have a relatively high coverage on the nine sub-cellular compartments. Following the redundancy removal as mentioned above, 2614 human (LOCATE _ human dataset) and 2690 mouse (LOCATE_mouse dataset) proteins in the two datasets were used as two of the independent datasets to test the performance of PreLocABC.
(4) The MultiLoc dataset was obtained from the supporting materials of Höglund, et al. [24] (http://www-bs.informatik.uni-tuebingen. de/Services/MultiLoc) and used as a test dataset of PreLocABC. The MultiLoc dataset was obtained by extracting all animal, fungal and plant protein sequences from the SWISS-PROT database, which contains a total of 5447 proteins in the nine sub-cellular compartments we chose. In order to obtain the independent test dataset, we also ran BLASTP in an all-on-all fashion for all sequences in this dataset by the same methods mentioned above to remove the redundancy between the training and MultiLoc datasets. Finally, we obtained a total of 4460 protein sequences (MultiLoc_4460 dataset) and used them as one of the independent test datasets for PreLocABC.
(5) The Cell dataset (1552 proteins) was obtained from the supporting materials of Foster et al. [5]. This study used PCP information, which is the quantitative data of all the proteins of a proteome separated into several consecutive cell fractions. This information was extracted and 621 proteins in the Cell dataset were assigned to one or more of nine possible sub-cellular localizations based on the annotation in SWISS-PROT. The rest of the proteins composed Cell_931 dataset which was predicted by the PCP-KNN sub-module. In order to obtain the high-quality training dataset, we filtered Cell_621 as follows. (i) We removed sequences with ambiguous and uncertain annotations such as 'by similarity', 'potential', 'probable', 'possible'. (ii) We clustered sequences at 80% identity using the cd-hit program [20] and removed highly homologous sequences from Cell dataset in order to eliminate the homologues and redundancy bias [13,21]. Finally, we got a total of 566 protein sequences and named the dataset as Cell_566. In order to measure the improvement of prediction efficiency when PCP-KNN sub-module was integrated into PreLocABC, the Cell_566 dataset was divided randomly into training and test datasets (Cell_566_train and Cell_566_test).
Table 1 lists the numbers of proteins for each sub-cellular compartment in training and test datasets for PreLocABC. Details of these datasets have been presented in Supporting materials\datasets.xls.
| Sub-cellular compartment | SP_human | SP_mouse | Hera_1124 | LOCATE_human | LOCATE_ mouse | MultiLoc | Cell_566 |
|---|---|---|---|---|---|---|---|
| cytoplasm | 1648 | 1335 | 138 | 409 | 607 | 1041 | 197 |
| endoplasmic reticulum | 298 | 448 | 253 | 109 | 169 | 168 | 104 |
| extracellular space | 742 | 719 | 118 | 164 | 164 | 663 | 44 |
| golgi apparatus | 225 | 386 | 58 | 104 | 146 | 116 | 50 |
| lysosome | 85 | 83 | 49 | 68 | 34 | 66 | 24 |
| mitochondria | 451 | 397 | 99 | 53 | 275 | 384 | 120 |
| nucleus | 1968 | 1336 | 258 | 1265 | 1055 | 766 | 62 |
| peroxisome | 58 | 53 | 14 | 17 | 37 | 127 | 19 |
| plasma membrane | 1243 | 1250 | 146 | 737 | 740 | 1129 | 41 |
Table 1: Numbers of proteins for every sub-cellular compartment in each datasets.
Construction of PreLocABC
Construction of five sub-modules integrated into PreLocABC: Five sub-modules integrated into PreLocABC were described in the following five sections:
(1) As described by Szafron et al. [18], each sequence was mapped into a set of features in the first sub-module. First, the query sequence was compared to the SWISS-PROT database using BLAST, and then the top three homolog SWISS-PROT entries (with E-values less than 0.001) were parsed to extract a feature set from the SWISS-PROT KEYWORDS field and the SUB-CELLULAR LOCALIZATION field. Next, the union of the feature set was used as input for the classification phases to predict the sub-cellular localization with a probability score. If no homolog matched the E-value cutoff or if no features were extracted from SWISS-PROT fields, no prediction was made.
(2) In the second sub-module, the average relative amino acid compositions (AACs) for proteins from each compartment were calculated for the N-terminal 25 residues (NTAAC) and the rest residues of the sequence (CTAAC) separately. And then the localization-specific domains were determined by comparing the occurrence patterns of domains across nine sub-cellular compartments from Pfam database (database of protein families, version 16.0), as described by Guda et al. [13]. This sub-module produced the final score based on the presence or absence of localization-specific Pfam domains in a given localization and the relative amino acid weights calculated from AAC.
(3) In the third sub-module, the protein features, such as sorting signals, amino acid composition and functional motifs were used to convert amino acid sequences into numerical vectors, which were then classified with a weighted KNN classifier, as described in Horton et al. [25]. The output indicated the number of nearest neighbors to the query sequence which localizes to each organelle.
(4) Most existing nuclear localization predictors were focused on discriminating statically nuclear proteins, rather than nuclear localization itself. Nuclear Localization Signals (NLS) were used in the fourth sub-module to identify proteins that are localized to the nucleus, either temporarily or permanently, based on a Support Vector Machine (SVM) with a custom kernel as shown in Hawkins et al. [10]. The output of this sub-module showed the probability of the query sequence to nuclear localization.
(5) In the fifth sub-module, N-terminal protein sorting signals were used as a feature to predict the localization sites by neural network according to the description of Emanuelsson et al. [8-9]. The output indicated how likely it was that the protein had a mitochondrial targeting peptide.
Construction of PreLocABC by integrating five sub-modules based on the bayesian model: In this step, PreLocABC integrated five sub-modules mentioned above to assign the likelihood to the nine compartments for each protein. First, the training dataset was used to train and calculate weight coefficients of five sub-modules and threshold values of each sub-cellular localization score. The sub-cellular compartments are denoted as follows:
C = {Ci}£-i = 1, 2, 3, 4, 5, 6, 7, 8, 9 (1)
where i refers to nine compartments: cytoplasm, ER, extracellular space, Golgi apparatus, lysosome, mitochondrion, nucleus, peroxisome and plasma membrane.
The five integrated sub-modules are denoted as follows:
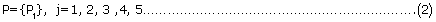
where j refers to the serial number of five sub-modules.
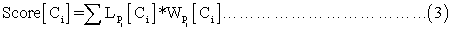
where 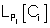 refers to the likelihood of a protein localized in Ci predicted by Pj and
refers to the likelihood of a protein localized in Ci predicted by Pj and 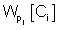 refers to the weight coefficients of likelihood in Ci predicted by Pj. There are 45 weights for 9 sub-cellular components of 5 sub-modules. Each weight received an initial value of 0.5. In each cycle, one weight was changed ranging from 0∼5 with step length 0.01 and the other 44 weights had no change. We chose the weight value when the prediction accuracy was the highest, and replaced the initial value of the weight with this training result in the next cycle.
refers to the weight coefficients of likelihood in Ci predicted by Pj. There are 45 weights for 9 sub-cellular components of 5 sub-modules. Each weight received an initial value of 0.5. In each cycle, one weight was changed ranging from 0∼5 with step length 0.01 and the other 44 weights had no change. We chose the weight value when the prediction accuracy was the highest, and replaced the initial value of the weight with this training result in the next cycle.
PreLocABC can predict the sub-cellular localization of query proteins by selecting the highest score for single-localized proteins or score threshold for multi-localized proteins. There are 9 score thresholds T[Ci] for 9 sub-cellular components in PreLocABC. Each score threshold received an initial value as the geometrical mean of scores for the corresponding sub-cellular component. In each cycle, one T[Ci] was changed ranging from the lowest score to the highest score with step length 0.01 and the other 8 score thresholds remained unchanged. We chose the threshold value when the accuracy of prediction was at the peak, and replaced the initial value of the threshold with this training result in the next cycle.
For single-localization, if
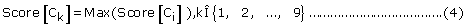
Then the set of protein sub-cellular localization predicted by PreLocABC can be defined as PLoc = Ck.
For multi-localization, if
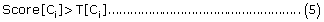
where T[Ci] refers to the score threshold corresponding to the highest prediction accuracy, then the set of protein sub-cellular localization predicted by PreLocABC can be defined as PLoc = {Ci}, i∈{1, 2,......., 9}.
Evaluation of the prediction performance
Independent data set test and ten-fold cross-validation were used to examine PreLocABC for its effectiveness in practical application.
Independent data set test: We used an independent data set test to evaluate the performance of PreLocABC we had built. The model is trained using the SP_human dataset and is tested on the remaining datasets (LOCATE_human, Hera, MultiLoc, LOCATE_mouse and SP_ mouse) which have been left untouched. None of the proteins in the independent test datasets occurs in the training dataset.
Ten-fold cross-validation: N-fold cross-validation not only provides an unbiased estimation of accuracy at much reduced computational cost, but is also considered an acceptable test for evaluating the prediction performance [26]. Therefore, in this study, 10-fold cross-validation was used to evaluate the performance of PreLocABC. In detail, the SP_human dataset was divided into 10 sets consisting of nearly an equal number of sequences. These 10 sets were further divided into training and test sets. The training and testing was carried out ten times at one particular value of weights and threshold, each time using nine sets for training and the remaining one set for testing. The final performance was obtained by averaging the performance of all the ten tests.
Evaluation parameters: PreLocABC is a novel predictor of protein sub-cellular localization for both single and multi-localized proteins. The evaluation parameters used by traditional predictors for singlelocalized proteins are not suitable for PreLocABC (the reasons have been shown in Supporting materials\supplement.doc\Section 1.1∼1.2). Therefore, we defined new Accuracy and MCC [27] as equation 6∼11 for the performance evaluation of PreLocABC (details on the definition of new evaluation parameters have been shown in Supporting materials\ supplement.doc\Section 1.3∼1.5).
The prediction performance of PreLocABC for each sub-cellular compartment:
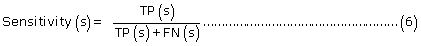
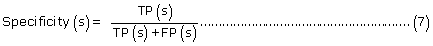
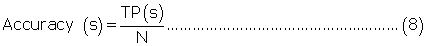
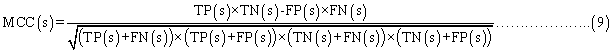
where TP(s), TN(s), FP(s) and FN(s) refer to the number of true positive, true negative, false positive and false negative predictions for each given sub-cellular compartment s, respectively.
The overall prediction performance of PreLocABC:
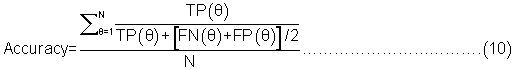
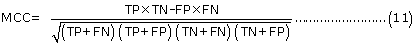
Where N refers to the total number of predicted proteins, θ refers to a given protein predicted by PreLocABC. 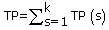 ,
, 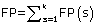 and
and 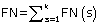 , where k refers to the number of sub-cellular compartments PreLocABC can discriminate.
, where k refers to the number of sub-cellular compartments PreLocABC can discriminate.
One application instance of PreLocABC in proteomics: the construction of PCP-KNN sub-module
In addition to integrating the five sub-modules mentioned above, we constructed another sub-module named PCP-KNN. It combines PCP method [2,5] based on the high-throughput mass spectrum quantitative information of proteins and fuzzy KNN algorithm [28] based on probabilistic classification.
Protein Correlation Profiling (PCP) method was a strategy to eliminate the noise caused by cross-organelle contamination in highthroughput mass spectrometry quantification of a proteome. To say specifically, in order to identify the localizations of a proteome, the cell lysate is firstly centrifugalized and separated into sequential gradient fractions, then proteins of each isolated organelle are identified and quantified by mass spectrometry. Due to cross-organelle contamination, a protein can be found in several or even all the fractions, however, proteins in the same organelle are supposed to share a consensus quantitative distribution pattern among consecutive fractions and we can utilize this phenomenon [2]. In Foster's study [5], the query protein's distribution curve was compared to a marker protein's distribution curve, and the set of quantitative distribution curve among consecutive fractions of each protein in a proteome was called the proteome's PCP data. Here we developed this method by comparing query protein with a cluster of marker proteins and calculating the probability that the query protein locates in each organelle by fuzzy K-Nearest Neighbors (fKNN) algorithm [28]. Fuzzy-KNN algorithm is more precise than basic KNN algorithm because it gives consideration to the continuous distance/similarity value between proteins and assigns a series of class membership of the query protein to each organelle rather than the crispy yes-or-no result of basic KNN algorithm.
Please see Supporting-materials\supplement.doc\Section 2 for more details of the procedure of PCP-KNN module and its results.
Statistical Analysis
A comparison of the prediction performance among PreLocABC, Proteome Analyst, pTARGET, WoLF PSORT and PCP-KNN was made using Fisher's exact test for any 2×2 tables and Pearson
x2 test for non- 2×2 tables by using SPSS13.0 [29]. Differences were considered to be statistically significant when the p value was less than 0.05.
Performance of PreLocABC from ten-fold cross-validation and independent data set test
In the 10-fold cross-validation, the average test accuracy was 79.39% and the average test MCC value was 0.78. The differences in the accuracy obtained from each test of the 10-fold cross-validation were of no statistical significance (x2=10.51, υ=9, 0.25
In the independent data set test, the accuracy for PreLocABC against human protein datasets Hera and LOCATE_human was 76.11% and 60.03%, respectively. In addition to human proteins, PreLocABC can also be used to determine the localization of proteins in other species. The test accuracy for PreLocABC performed on the mouse protein datasets SP_mouse and LOCATE_mouse, and mixed species protein dataset MultiLoc was 76.26%, 57.96% and 76.79%, respectively.
The MCC [30,31] values are usually employed while evaluating the performance on unbalanced datasets. They can range from -1 to +1 and higher values indicate better predictions, considering both the true positives and the true negatives as successful predictions. Therefore, in addition to the overall accuracy, the MCC values were also tested due to the imbalance of numbers of proteins localized in different compartments, such as 1648 of cytoplasm vs. 28 of peroxisome in SP_human data set (Table 1). The MCC values of PreLocABC were 0.71, 0.56, 0.72, 0.55 and 0.72 for Hera, LOCATE_human, SP_mouse, LOCATE_mouse and MultiLoc datasets , respectively.
Figure 1 presents the detailed results for individual compartments. The highest accuracy and MCC values were obtained when a query protein was localized to the nucleus for human proteins, and to the lysosome for mouse proteins.

Figure 1: Sub-cellular compartmental prediction accuracy (A) and MCC (B) of PreLocABC against five different test datasets: Locate_human dataset, Hera dataset, MultiLoc dataset, Locate_mouse dataset and SWISS-PROT mouse dataset (please see details in Section 2.1). ‘mi’ refers to mitochondrion, ‘cy’ refers to cytoplasm, ‘er’ refers to endoplasmic reticulum, ‘ex’ refers to extracellular space, ‘go’ refers to Golgi apparatus, ‘ly’ refers to lysosome, ‘nu’ refers to nucleus, ‘pe’ refers to peroxisome and ‘pm’ refers to plasma membrane. The red arrows refer to the highest accuracy and MCC values for human datasets and mouse datasets, respectively.
Comparison of PreLocABC with other existing predictors
We compared the performance of PreLocABC with other three existing efficient predictors: Proteome Analyst, pTARGET and WoLF PSORT across the same five different datasets [Hera, LOCATE_human, LOCATE_mouse, MultiLoc and SP_mouse, please see section 2.1- (1∼4) for details of these datasets]. Among these predictors, Proteome Analyst and WoLF PSORT can predict the localization site(s) of both single-localized and multi-localized proteins.
From Table 2 and Figure 2, we find that PreLocABC outperformed other subcellular localization methods. First, the overall prediction accuracy of PreLocABC was significantly higher than that of any other three existing algorithm against all five independent datasets (P<0.005). Especially for LOCATE_human and LOCATE_mouse datasets, the overall prediction accuracy of PreLocABC was ∼20%, ∼25% and ∼50% higher than that of Proteome Analyst, pTARGET and WoLF PSORT, respectively (Table 2). Second, the prediction accuracy of PreLocABC for Golgi apparatus proteins was significantly higher than that of any other three existing predictors (P<0.05) against five independent datasets (Figure 2). Third, despite the low number of lysosomeslocalized and peroxisome-localized proteins, these localizations can be well predicted by PreLocABC with two or three independent datasets, compared with the three existing predictors (P<0.05, Figure 2). Fourth, prediction performance of PreLocABC on endoplasmic reticulum, mitochondrion, extracellular region and cytoplasm is also better than that of other existing predictors for two or three independent datasets (P<0.05, Figure 2).
| Algorithm | LOCATE_human | Hera | MultiLoc* | LOCATE_mouse | SP_mouse |
|---|---|---|---|---|---|
| Accuracy (%) | |||||
| PreLocABC | 60.03** | 76.11** | 76.79** | 57.96** | 76.26** |
| Proteome Analyst | 48.17 | 72.8 | 70.15 | 46.24 | 63.33 |
| pTARGET | 45.13 | 66.09 | 70.44 | 44.53 | 57.67 |
| WoLF PSORT | 30.19 | 60.08 | 62.93 | 30.25 | 49.86 |
| MCC | |||||
| PreLocABC | 0.56 | 0.71 | 0.72 | 0.55 | 0.72 |
| Proteome Analyst | 0.47 | 0.7 | 0.68 | 0.46 | 0.64 |
| pTARGET | 0.38 | 0.61 | 0.66 | 0.37 | 0.51 |
| WoLF PSORT | 0.3 | 0.59 | 0.59 | 0.29 | 0.45 |
‘*’ The difference in the overall accuracy of Proteome Analyst and pTARGET
for MultiLoc dataset had no statistic significance. Therefore, we performed the
comparison of the overall accuracy among PreLocABC, pTARGET, and WoLF
PSORT using Pearson x2 test (ν=2) by SPSS13.0.
‘**’ refers to the comparison of the overall accuracy of PreLocABC with other
existing predictors, p<0.005
Table 2: All-Accuracy (%) and All-MCC value of PreLocABC against different independent datasets.

Figure 2: MCC values and prediction accuracy of different predictors for nine sub-cellular compartments based on five test datasets: Locate_human dataset (A and B), Hera dataset (C and D), MultiLoc dataset (E and F), Locate_mouse dataset (G and H) and SWISS-PROT mouse dataset (I and J) (please see details in Section 2.1). PA refers to Proteome Analyst. WP refers to WoLF PSORT. ‘mi’ refers to mitochondrion, ‘cy’ refers to cytoplasm, ‘er’ refers to endoplasmic reticulum, ‘ex’ refers to extracellular space, ‘go’ refers to Golgi apparatus, ‘ly’ refers to lysosome, ‘nu’ refers to nucleus, ‘pe’ refers to peroxisome and ‘pm’ refers to plasma membrane. ‘*’ refers to the comparison of the prediction accuracy of PreLocABC with other existing predictors, p<0.05.
One application instance of PreLocABC in proteomics: the construction of PCP-KNN sub-module
In order to extend the application of PreLocABC, we integrated the PCP-KNN sub-module, the input of which was proteome massspectrum quantitative data on consecutive cell fractions. Here, we divided Cell_566 dataset which contains proteins with both location information from SWISS-PROT and PCP information from Foster's experiment [5] randomly into the training and test sets [please see section 2.1-(5) for details]. After the integration with the PCP-KNN sub-module, the overall accuracy of PreLocABC was significantly increased (P<0.005), so was the trend of MCC value. Especially, its specificity and sensitivity for predicting extracellular space-localized, nucleus-localized, cytoplasm-localized and plasma membranelocalized proteins were enhanced remarkably (Figure 3).

Figure 3: All-MCC (A), All-Accuracy (B), specificity (C) and sensitivity (D) of different predictors tested using the independent dataset. ‘**’ refers to the comparison of the All-Accuracy between PreLocABC and PCP-KNN, p<0.01; ‘*’ refers to the comparison of All-Accuracy of PreLocABC before and after the integration with PCPKNN, p<0.05. ‘mi’ refers to mitochondrion, ‘cy’ refers to cytoplasm, ‘er’ refers to endoplasmic reticulum, ‘ex’ refers to extracellular space, ‘go’ refers to Golgi apparatus, ‘ly’ refers to lysosome, ‘nu’ refers to nucleus, ‘pe’ refers to peroxisome and ‘pm’ refers to plasma membrane. The red underlines refer to the higher specificity and sensitivity of PreLocABC after being integrated with the mass-spectrum quantitative information of proteins than before.
Given the importance of protein sub-cellular localization, a large number of computational predictive algorithms have been developed by different machine learning methods that take into account various protein characteristics. However, it remains highly challenging to improve the performance of the existing predictors for protein subcellular localizations on a large scale. Here, we introduce a novel protein sub-cellular localization predictor-PreLocABC, which integrated five sub-modules based on different features of a protein by a Bayesian model, to calculate the likelihood of a query protein belonging to a particular sub-cellular compartment.
Compared with the existing predictors, PreLocABC has several advantages as follows.
First, PreLocABC integrates more localization features of proteins, such as homology, amino acid composition, sorting signals and functional motifs than most of existing predictors (Details of the characteristics of the existing predictors for protein subcellular localization have been summarized in Supporting materials\ supplement.doc\Table S1).
Second, PreLocABC can predict more sub-cellular locations than many other predictors. According to literature, there have been several approaches integrating pre-existing predictors. For example, Liu et al. [32] integrated 12 predictors from eight independent sub-cellular localization predicting programs. However, they can only perform four-compartment eukaryotic sub-cellular localizations prediction (nuclear, cytoplasmic, mitochondrial and extracellular) and has no web server; Shen et al. [33] integrated 9 predictors, but they can only predict whether or not a protein is localized in mitochondrion.
Third, in the comparison of the performance relative to individual sub-cellular localization, PreLocABC has higher prediction reliability and efficiency for many sub-cellular compartmental proteins, even than Proteome Analyst which has been considered the best predictor in terms of sensitivity and specificity to different sub-cellular compartments [34], which might be because proteins in the independent test datasets of this study have low homology with the training sets of Proteome Analyst. In addition, despite the low number of Golgi apparatus, lysosomes and peroxisome proteins, these localizations can be better predicted by PreLocABC than the existing predictors on more than two independent data sets (Figure 2).
Fourth, PreLocABC can predict not only single-localized proteins, but also multi-localized ones. There are a considerable number of proteins targeting to multiple compartments, shuttling between compartments or localizing to the boundary of different organelles. In PreLocABC, protein locations can be identified by testing whether the score is above the threshold of each compartment. Compared with two existing predictors (Proteome Analyst and WoLF PSORT) which can also predict the sub-cellular localization of both single-localized and multi-localized proteins, PreLocABC has better performance (Table 2). Moreover, the first well-founded definition of overall prediction accuracy for the multi-compartmental prediction was constructed in this study. Compared with the previous literature of the multicompartmental predictors [35,36], it may be able to find the proper threshold based on the best accuracy and be used to evaluate the performance of the multi-compartmental predictors more reasonably (please see section 2.3 and Supporting materials\supplement.doc\ Section 1 for details).
Finally, PreLocABC is not only a predictor of protein sub-cellular localization, but also an integration strategy, which can integrate different localization features as needed. Current high-throughput sub-cellular proteomics technology has been used to map the subcellular localization of a number of proteins in different organs [37,38]. However, due to the high sensitivity of mass spectrometers and the difficulties inherent in purifying organelles to homogeneity, it is hard to distinguish bona fide proteins from those contaminators. As an application instance of PreLocABC , we constructed PCP-KNN sub-module to integrate the proteomics mass-spectrum quantitative information of query proteins into PreLocABC and its prediction performance was improved remarkably (Figure 3), which indicates that this integration strategy can utilize more and more localization-related features of proteins and enhance the prediction performance effectively.
Furthermore, there are two matters of interest that are worthy of discussion. First, the five independent test sets in this study are derived from SWISS-PROT database, LOCATE database and MultiLoc dataset, which have been used to evaluate the performance of several previous predictors [32,34,39]. In the independent dataset test of PreLocABC and performance comparison of PreLocABC with the existing predictors, we found that all of the sub-cellular localization predictors showed a lower level of sensitivity when applied to LOCATE_human and LOCATE_mouse datasets. The sub-cellular localization data within LOCATE database were determined by a high-throughput, immunofluorescence-based assay and by manually reviewing peerreviewed publications [23], but not with inclusion of information from other sources, including SWISS-PROT. It therefore represents a suitable independent evaluation set that will have less overlap with the training sets originally used to develop the sub-cellular localization predictors [34]. Thus the predictors, such as Proteome Analyst, that incorporate homology searches on SWISS-PROT showed the largest decrease in overall performance, whereas PreLocABC had the lowest reduction when applied to LOCATE_human and LOCATE_mouse datasets (Table 2). Second, some protein features, such as amino acid composition, are used by more than two sub-modules integrated by PreLocABC. This Figure 3: All-MCC (A), All-Accuracy (B), specificity (C) and sensitivity (D) of different predictors tested using the independent dataset. '**' refers to the comparison of the All-Accuracy between PreLocABC and PCP-KNN, p<0.01; '*' refers to the comparison of All-Accuracy of PreLocABC before and after the integration with PCPKNN, p<0.05. 'mi' refers to mitochondrion, 'cy' refers to cytoplasm, 'er' refers to endoplasmic reticulum, 'ex' refers to extracellular space, 'go' refers to Golgi apparatus, 'ly' refers to lysosome, 'nu' refers to nucleus, 'pe' refers to peroxisome and 'pm' refers to plasma membrane. The red underlines refer to the higher specificity and sensitivity of PreLocABC after being integrated with the mass-spectrum quantitative information of proteins than before. may inevitably affect the independence of the different sub-modules. The protein features which are used repeatedly may play key roles in the determination of protein sub-cellular localization. Moreover, the result of this study also demonstrates that the prediction performance of PreLocABC has been improved by integrating various sub-modules.
In conclusion, PreLocABC as a novel predictor for protein subcellular localization is presented here, covering nine major localizations. An interesting characteristic of the present method is the integration of different protein features as needed, which supports the assignment of the protein sub-cellular localization more reliably and with high accuracy. Although the current version of PreLocABC package can cover 9 sub-cellular location sites of animal proteins, which far outnumber three or four location sites covered by several existing predictors [40-42], if a query protein is outside of the 9 location sites, the predicted result would still make no sense. A similar limitation in the coverage scope also exists for other Web-server predictors [43]. In view of this, as more experimental sub-cellular localization data become available, we will periodically expand the coverage scope for the Web servers in the future version of PreLocABC. In a word, PreLocABC is able to complement the existing sub-cellular localization predictors and provides an alternative way for biologists to predict protein sub-cellular localization.
We thank Xiaohong Qian, Wantao Ying, Songfeng Wu, Yunwei Hao, Aihua Sun and Lin Hou in Beijing Proteome Research Center for their helpful discussion; our IT Engineer Yi Liu for publishing the online predicting tool. We also thank two anonymous reviewers for their helpful comments. This work was supported by the Chinese National Key Program of Basic Research [2006CB910803, 2006CB910706, 2010CB912700, 2011CB910600], the National High Technology Research and Development Program of China [2006AA02A312], National Science and Technology Major Project [2008ZX10002-016, 2009ZX09301-002], National Natural Science Foundation of China [30800200] and State Key Laboratory of Proteomics [SKLP-Y200811, SKLP-K200906].