Translational Medicine
Open Access
ISSN: 2161-1025
ISSN: 2161-1025
Review Article - (2012) Volume 0, Issue 0
Chronic myeloid leukemia (CML) is a chronic myeloproliferative disorder with leukemic cells featuring the Philadelphia (Ph1) chromosome comprising the reciprocal chromosomal translocation t(9;22)(q34;q22) and the resultant constitutive active Bcr-Abl tyrosine kinase (TK). The introduction of disease-specific molecular targeted agents, that is, TK inhibitors (TKIs) for Bcr-Abl TK, such as the first-in-class TKI imatinib mesylate (IM) or the secondgeneration TKIs (SGIs) nilotinib and dasatinib, has dramatically improved the long-term treatment outcome for CML in this century. In addition, several new SGIs, such as bosutinib or bafetinib, are under development, while ponatinib, which is active against CML refractory for all preceding TKIs, for example, CML with T315I Abl kinase domain mutation, is currently being evaluated in clinical trials. Because those TKIs have different sensitivity for Bcr- Abl mutation and profiles for adverse effects, a thorough understanding of the pharmacologic characteristics of TKIs is mandatory for their safe and effective clinical use. Recent studies have clearly shown the faster and deeper responses to SGIs, both nilotinib and dasatinib, compared with those to IM, indicating the need for a paradigm shift in approaches to TKI therapy for treatment-naïve CML. In addition, evidence accumulated during the past decade has indicated optimal methodologies for monitoring the treatment effect of TKIs, selecting the appropriate therapeutic strategies, and predicting the outcome for treatment with TKIs for individual patients with CML. In this article, we review the principles and current knowledge and topics of the various uses of TKIs for CML. We also touch upon the reason why the faster and the deeper responses to TKIs is the prerequisite for their safer use and longer-lasting positive treatment outcome for CML.
<Keywords: Chronic myelogenous leukemia, Tyrosine kinase inhibitor, Imatinib, Nilotinib, Dasatinib
Chronic myeloid leukemia (CML) is a subtype of chronic myeloproliferative disorders with leukemic cells featuring the reciprocal chromosomal translocation t(9;22)(q34;q22), also known as the Philadelphia (Ph1) chromosome, and its resultant Bcr-Abl fusion oncoprotein, which exerts constitutive tyrosine kinase (TK) activity which in turn stimulates various downstream signaling cascades for deregulated cell proliferation, cell survival, resistance to cytotoxic stimuli and genetic instability [1,2]. Until a decade ago, long-term disease control was achieved only by allogeneic hematopoietic stem cell transplantation (HSCT) with a complete cure rate of about 50- 60%, while the effects on CML of pharmacologic interventions, such as the use of interferon-α (IFN-α), were rather limited [2]. However, the introduction of tyrosine kinase inhibitors (TKIs) for Bcr-Abl TK, such as the first-in-class TKI imatinib mesylate (IM), as disease-specific molecular targeted agents has dramatically improved the long-term treatment outcome for CML in this century. The IRIS (International Randomized Study of Interferon and STI571) study of chronic phase CML (CML-CP) patients which compared the effects of IM and IFN-α plus cytarabine as first-line therapy found that continuous IM treatment resulted in greater treatment success with an eight-year overall survival (OS) rate of 93% and cumulative complete cytogenetic response (CCyR) of 83%, thus demonstrating the apparent advantage of firstline IM therapy for better long-term outcomes for CML patients [3,4]. Despite these excellent results, caution is needed when interpretating the findings of this study because the analysis was based on the data for patients who successfully continued IM treatment. Indeed, subsequent reports brought out the fact that approximately 30-40% of the patients initially enrolled in the IM cohort of the IRIS study dropped out from the trial due to unsatisfactory or no response or to intolerance for IM. Moreover, even among the patients whose data were included in the analysis, 17% did not attain CCyR and 10% showed disease progression [5]. In view of these findings, comparison of much longer-term effects, e.g., over at least 20~30 years, to determine long-term outcomes for IM and allogeneic HSCT remain a matter for future studies. Nevertheless, the apparently better OS rate and the notably fewer short-term lifethreatening adverse events attained with IM in comparison with allogeneic HSCT, the former has come to be regarded as the first-line treatment for CML, especially in the chronic phase (CP). Moreover, the development of second-generation TKIs (SGIs), such as dasatinib and nilotinib, which can overcome IM resistance and intolerance, has been a topic of heated debate concerning the treatment for CML during the past decade. Moreover, even newer agents, such as bosutinib, bafetinib and ponatinib, are currently being developed [6,7].
In the clinical setting, an in-depth understanding of the pharmacologic characteristics of each TKI, including its kinase inhibitory ability, affinitiy for mutated Abl, as well as adverse effects and predictors for clinical outcomes, is essential for choosing the most appropriate agent for individual CML patients. This article reviews the current knowledge which is essential for the effective use of Bcr-Abl TKIs and focuses especially on the effects and limitations of IM and SGIs for CML.
Positive and Negative Aspects of the use of IM for CML
The first report regarding the dramatic preclinical effect of CGP57148 (later renamed STI571 and then IM), an ATP-competitive TKI which is mostly specific for the Abl protein tyrosine kinase in Bcr-Abl-positive leukemias was announced by Dr. B.J. Druker in 1996 [8]. IM inhibits the constitutive active status of autophosphorylated Abl by binding to the ATP-binding site of the inactive conformation of Bcr-Abl in which the kinase-active site is shielded by the activation loop (A-loop) [9], resulting in blocking of Bcr-Abl TK-mediated downstream signaling for leukemogenesis. In addition to Bcr-Abl TK, IM also inhibits, and even more strongly, c-KIT and PDGF-R [10,11]. As a consequence, IM inhibits cell proliferation and induces programmed cell death mainly via apoptosis employing Bim and Bad as the crucial pro-apoptotic BH3-only proteins [12].
Since the introduction of IM, its development as a therapeutic agent has progressed very quickly, and within a couple of years IM gained its position as the first-line therapeutic pharmacologic intervention. Besides its major benefits for CML patients, the successful development has provided new insights for a better understanding of the disease biology. Also, among the many scientific contributions made by the introduction of IM, the major progress made in research in identification of the molecular mechanisms involved in TKI resistance has provided numerous scientific insights into the development of new molecular targeted agents for CML as well as other types of cancerous diseases. However, it was found that the mechanisms for resistance against IM are different from those against conventional genotoxic agents. As early as one year after the clinical approval of IM for CML, Sawyer’s group identified two major mechanisms for IM resistance, T315I genomic point mutation in Abl kinase domain (KD), which results in the conformational change of the IM binding site, and bcrabl gene amplification, which results in Bcr-Abl overexpression [13]. Soon after those epoch-making discoveries, other mechanisms for IM resistance, such as various types of Abl KD mutations [14,15], Bcr-Abl overexpression due to transcriptional upregulation or the activation of alternative kinases such as Lyn, which promotes leukemic cell proliferation and survival, were also identified [16,17]. At present, more than 100 types of Abl KD mutations have been identified as causative for modest to complete resistance against IM, and it has been demonstrated that one or more clones with mutated Abl KD may be present in approximately half of all IM-resistant CML patients. The underlying molecular mechanisms for IM resistance vary depending on the type of Abl KD mutation. For instance, T315 is essential for the hydrogen bond between IM and Abl KD, so that mutations in T315, such as T315I or T351A, result in interference with this action. Moreover, mutations in T315 lead to conformational changes in adjunctive amino acids such as K271, E286M, R362, or D381, which are also crucial for IM-binding to Abl KD. Among mutations in the phosphate-binding loop (P-loop), those at Y253, such as Y253H, also disrupt the hydrogen bond between IM and Y253, while other types of P-loop mutations, such as G250E or E255K, stabilize Bcr-Abl as an active form which is insensitive to IM. Mutations in the catalytic loop (C-loop), such as M351T or E355G, also cause conformational changes in an inactive form of Abl, thereby preventing the binding of IM. More recent studies have also identified other Bcr-Abl-unrelated mechanisms for TKI resistance, such as abnormal influx/efflux pump expression [18,19], multifactorial support by bone marrow microenvironment [20-22], deregulation of programmed cell death machineries [12,23-25], or drug insensitivity due to leukemic stem cell phenotype [26-28]. All these molecular mechanisms combine to promote IM resistance.
The other problem is adverse events caused by IM, which are most likely associated with its inhibitory effect on off-target kinases. Hematological adverse events, one of the most frequent complications of IM treatment, may be associated with the inhibitory effect of IM on c-KIT, while some of the non-hematologic adverse events, such as skin rash, myalgia, edema, or fluid retention, may be associated with its inhibitory effect on PDGF-R. In the IRIS study, 8% of the patients dropped out from the study during the 6-year follow-up due to intolerance to IM [5].
Development of sgis and their characteristics
To overcome the major mechanisms for IM resistance, several SGIs with more flexiblity in response to Abl KD conformational changes, higher affinity for more abundant Bcr-Abl and ability to block various leukemogenic kinases have been developed for or introduced to the treatment for CML. As of this writing, nilotinib and dasatinib have been approved for clinical use not only as second-line but also as first-line treatment, while bosutinib, bafetinib and ponatinib are undergoing clinical trials. As shown in Table 1, all SGIs possess much higher affinity for wild type Bcr-Abl TK [29-33]. While nilotinib potently inhibits the inactive form of Bcr-Abl, other SGIs are potently affect both active and inactive forms of Bcr-Abl [34]. Nilotinib is more highly selective for Bcr-Abl, and its effects on c-KIT and PDGF-R are much weaker than that on Bcr-Abl, so that it can be expected that its adverse events due to the inhibition of off-target kinases will be far less frequent than those associated with IM. While dasatinib and bosutinib exert multikinase inhibitory effects and bafetinib potently inhibits Lyn, a member of the Src kinase proteins, bosutinib does not inhibit c-KIT. Whether this insensitivity of bosutinib for c-KIT influences its anti- CML effect has not been clarified, but caution is advisable because the findings of several basic studies suggest that the concomitant blockade of Bcr-Abl and c-KIT is essential for suppressing Bcr-Abl-positive leukemias [35,36]. All SGIs possess different affinity (in vitro kinase inhibitory) profiles for mutated Abl KD. For instance, nilotinib is not effective for P-loop mutations and dasatinib not for F317 mutations [37]. Importantly, none of the TKIs except ponatinib is at all potent in inhibiting T315I, while the newest TKI ponatinib has been developed to overcome T315I mutated Bcr-Abl and has shown favorable clinical effects for CML patients who were resistant to previous TKIs regardless of the presence of T315I mutation. Thus, appropriate choice of an agent according to the type of Abl KD mutation is mandatory, especially when it is to be used as the second-line treatment for IM-resistant CML [38].
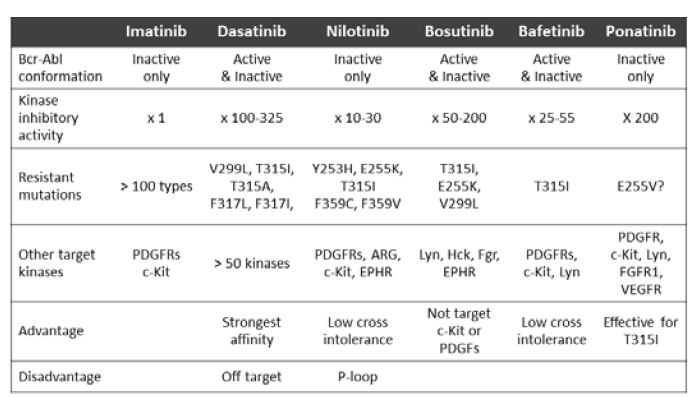
Table 1: Characteristics of tyrosine kinase inhibitors for chronic myelogenous leukemia.
Milestone for IM treatment of treatment-naïve CM
Accumulating evidence has provided clues for information relevant for predicting long-term outcome of IM treatment as first-line treatment for CML. The European LeukemiaNet (ELN) has established guidelines for IM therapy for patients with newly diagnosed CMLCP, which make it possible to judge whether the continuation of IM treatment is optimal for individual patients at several time points during the treatment course [39]. These guidelines proposed in 2006 classify the response status into ”Optimal”, “Suboptimal” and “Failure” for individual patients according to the degree of response to IM, i.e., hematologic response (HR), cytogenetic response (CyR), and molecular response (MR) at 3, 6, 12 and 18 months of IM treatment (Table 2). In addition to these three classifications, the criterion “Warnings” is suggested for patients showing any additional chromosomal abnormalities in Philadelphia-positive leukemic cells at diagnosis, an increase in bcr-abl copy numbers at any time point, or the emergence of clones with abnormal karyotype in non-Philadelphia hematopoietic cells. More recently, the revised guidelines proposed in 2009 include an evaluation system for the effect of SGIs on IM-resistant CML-CP. In these revised guidelines, the response status has been classified into “Suboptimal”, “Failure” and “Warnings”, while “Optimal” was not included due to the lack of convincing evidence for the long-term effects of SGIs on IM-resistant patients [40]. Indeed, although promising as salvage therapy, the effects of SGIs on IM-resistant CML-CP patients have been reported to be much inferior to those on treatment-naïve CML-CP cases [41,42]. According to these guidelines, patients who do not achieve any CyR at 3 months, partial CyR (PCyR) at 6 months, or major molecular response (MMR) at 12 months are evaluated as “Failure”.
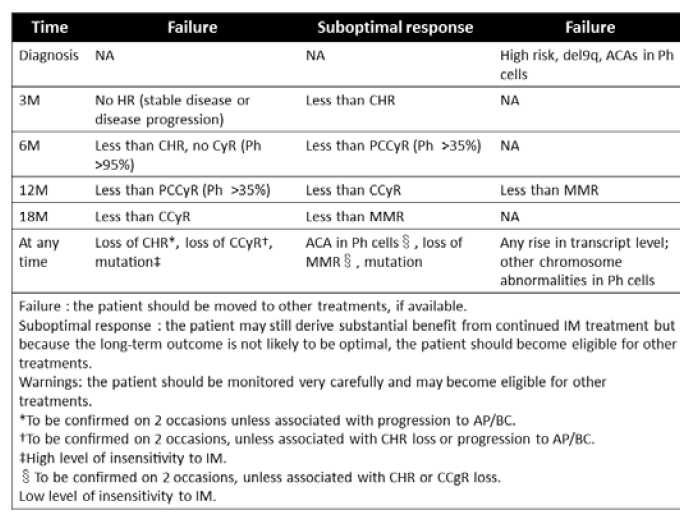
Table 2: European LeukemiaNet consensus guidelines for imatinib treatment for treatment-naïve chronic myelogenous leukemia.
The faster and deeper the response, the better the outcome may be
In brief, the ELN consensus guidelines propose that the faster and the deeper the response to IM treatment is, the more favorable the prediction is for long-term disease control of CML. In contrast, a brief look at the results of the IRIS study, which evidenced a continuous increase in the study population with cumulative incidence of CCyR as the observation period became longer, may justify expectations for the growing presence of late responders to continuous IM treatment. Indeed, in the pre-SGI era, we continued IM treatment, unless there was evidence of disease progression, even for so-called “non-optimal” responders in the hope they would turn out to be “late responders”. However, several studies have clearly shown the risk for late responders to IM by demonstrating that, according to Quintas-Cardama et al., late responders to IM are major candidates for future loss of response to IM [43]. These researchers revealed that, when compared with patients who achieved CCyR within 12 months of IM treatment, those who attained CCyR later were at significantly higher risks of disease progression, such as the loss of CCyR, after 60 months of treatment [43]. Similarly, de Lavallade etal. reported that failure to achieve CCyR after 12 months of IM treatment is significantly associated with later disease progression, especially after 48 months of treatment with IM. In addition to the timing of response, the depth of response is also strongly associated with long-term outcome [44]. Even for patients with CCyR within 12 to 18 months of treatment with IM, attainment of major molecular response (MMR) was significantly associated with longer maintenance of CCyR [45]. Hughes et al. stressed the importance of earlier attainment of MMR with IM treatment, showing that patients with bcr-abl transcripts > 10% at 6 months and > 1% at 12 months had inferior EFS and higher rate of disease progression. Their study also showed that patients who attained MMR by 18 months enjoyed markedly long-lasting responses without disease progression and with 95% EFS at 7 years [46].
To identify the basis of the need for faster and deeper effect of TKIs for patients with CML-CP, we need to briefly review the natural history of CML. CML starts with the acquisition of clonal proliferative property by acquiring Bcr-Abl TK activity in the very early phase of the disease (early CP). This is followed during the next 5 to 10 years by various additional chromosomal and oncogenic changes unrelated to Bcr-Abl, which have a cumulative effect in a multistep manner due to genetic instability caused by Bcr-Abl TK. During this process, leukemic cells, which initially show strong addiction to Bcr-Abl TK activity for their proliferation and survival, undergo a clonal evolution and loose their dependence on Bcr-Abl TK. This evolution then results in disease progression from CP to the advanced phases, that is, the accelerated and blast crisis phases (AP and BC) [47,48]. The more and the longer residual Bcr-Abl-positive leukemic cells survive during TKI treatment, the more frequently mutations for clonal evolution are likely to occur. Once leukemic cells that are less addicted to Bcr-Abl TK emerge, they may at first create chimeras with Bcr-Abl TK-dependent clones, but later develop into major clones during TKI treatment, and eventually cause the TKI treatment to fail. In view of this scenario, it makes sense to try and reduce the chance for clonal evolution by suppressing Bcr- Abl-positive clones as much and as fast as possible with TKIs to achieve the so-called “safe haven” status. The advisability of this strategy is supported by the fact that the maintenance of CCyR for more than 3 years is associated with the longer event free survival (EFS) and a reduction in the occurrence of future adverse events from 5.4% to 0.3% [5].
SGIs as first-line and second-line treatment
On the basis of the principle that the faster and deeper the response, the better the outcome and the greater the pharmacological potency may be, how can we best take advantage of the potency of SGIs? This question may already have been answered by the findings of two large prospective trials, ENESTnd (Evaluating nilotinib efficacy and safety in clinical trials - newly diagnosed patients), which conducted a head-tohead comparison of the effects of nilitinib and IM [49] and DASISION (Dasatinib versus imatinib study in treatment-naïve CML), which conducted a head-to-head comparison of the effects of dasatinib and IM for treatment-naïve CML-CP [50]. Both studies clearly showed the faster and deeper effects of SGIs in terms of achievement of CCyR and MMR, while SGIs induced complete MR (CMR) in more patients. After 18 months of treatment, nilotinib and dasatinib had induced CMR in 21% and 13% of patients, respectively, while IM had induced CMR in only 6 to 7%. Similar results have been reported by trials conducted by GIMEMA (Gruppo Italiano Malattie Ematologiche dell’Adulto) or the M.D. Anderson Cancer Center [51]. Importantly, these studies indicated that the faster and the deeper the effects of SGIs were, the more they eventually resulted in less disease progression and longer OS periods. In the ENESTnd trial, disease progression was observed in 1/283 patients and the 12-month OS ratio was 99.3% for the nilotinib cohort, while the corresponding findings for the IM cohort were 11/283 patients and 96.9%. In the DASISION trial, disease progression at 12 months was observed in 5/269 patients in the dasatinib cohort and in 9/260 patients in the IM cohort. Further, the rates for drug cessation due to intolerance were not significantly different for IM and nilotinib or dasatinib. These data constitute sufficiently supportive evidence for the use of nilotinib and dasatinib as the first-line treatment for CML-CP. In contrast, the utility of bosutinib as the first-line treatment for CML-CP remains controversial. In the BELA (Bosutinib efficacy and safety in newly diagnosed chronic myeloid leukemia) trial, achievement of CCyR with bosutinib was not significantly superior to that with IM at 12 months (70% vs 68%), although the achievement of MMR was better for the bosutinib arm than for the IM arm (39% vs 26%). Moreover, drug cessation due to adverse events, such as severe diarrhea, was notably more frequent for the bosutinib arm than for the IM arm (19% vs 5%) [51].
In contrast to the findings for nilotinib and dasatinib as first-line treatment, their use as second-line treatment following IM treatment was not as effective. According to the results of START-C trial, after 2 months of treatment with dasatinib, CCyR and MMR had been achieved in, respectively, 45% and 37% of IM-resistant, and 78% and 78% of IM-intolerant patients with CML-CP [42]. These effects were much inferior to those obtained for treatment-naïve CML-CP, and, as was to be expected, the effects of dasatinib were rather limited for advanced phase CML [52-54]. Similarly, the effect of nilotinib as second-line treatment is also much inferior to that observed in treatment-naïve patients. The CCyR rates after 24 months of treatment with nilotinib for IM-resistant and IM-intolerant CML-CP patients were reportedly 41% and 51%, respectively, while the MMR rate obtained with nilotinib for these patients was as high as 28% [55]. These results imply that there is a need for predictors for response to second-line SGIs for the development of alternative treatment strategies.
Current topics concerning the use of TKIs based on 10-year experience
Accumulating evidence has yielded important information for ways of monitoring treatment response and choice of treatment but also have given rise to new clinical questions. The next 7 sections deal with several topics regarding the use of TKIs based on a decade of experience.
1. Optimal timing for the switch from IM to sgis: It may be easy to decide this switch in the case of primary “Failure” for treatment with IM according to the ENL guidelines. The switch may be also not so difficult in case of intolerance IM, because cross-intolerance between IM and SGIs occurs infrequently [56,57]. The question that remains is the optimal timing for the switch from IM to SGIs in case of the loss of response in patients with initial optimal response. Kantarjian et al. reported that the effect of dasatinib as the secondline treatment for patients who have lost response to IM is affected by the timing of the drug switch. They classified patients into two groups according to whether they received dasatinib after the loss of major CyR (MCyR) or after the loss of McyR with concomitant loss of hematologic response (HR) when treated with IM. Although second-line dasatinib induced similar rates of complete HR (CHR) in both groups, it induced CCyR in 72% of the first and in 42% of the second group, and 2-year progression-free survival (PFS) of 89% and 29%, respectively. These trends were not influenced by the presence of Abl KD mutations except for that of T315I [58]. These results suggest that an earlier treatment switch is recommended, at least for patients who experienced secondary “Failure” for IM.
2. Increase in bcr-abl copy numbers and emergence of Abl KD mutated clone despite sustained ccyr: There is a high risk of failure to achieve CCyR with TKI treatment is with the occurrence of certain events, such as an increase in bcr-abl copy numbers, emergence of Abl KD mutation and the eventual disease progression. What exactly are the effects of such events in spite of sustained CCyR? Studies so far have suggested that an elevation of bcr-abl copy numbers, including the loss of MMR, during TKI treatment is frequently associated with the emergence of a clone with mutated Abl KD even in patients with CCyR. Kantarjian et al. reported that 116 (including 40 without MMR) of 258 patients with CCyR showed definite elevation of bcrabl copy numbers, and that 48 of the 76 patients with MMR lost it during IM treatment. Importantly, disease progression was observed only when the increase in bcr-abl copy number exceeded the threshold level of MMR. In contrast, the loss of MMR was found to be associated with disease progression in 6/48 patients, even though these patients maintained their CCyR [59]. It is therefore important to assess the presence of Abl KD mutation and to consider a switch to different TKIs when bcr-abl copy numbers increase, especially in case of loss of MMR.
3. Biomarkers for clinical outcome of treatment-naïve CML in TKI treatment (except Abl KD mutations and bcr-abl copy numbers): Several biomarkers have been identified to be predictive for the clinical outcome of treatment-naïve CML patients in TKI treatment. While the Sokal score was originally established as a prediction model to predict survival of patients with conventional chemotherapies, IRIS study revealed a correlation between Sokal score and response to IM [3]. Some in vitro assays are also informative for the prediction of outcome of CML in TKI treatment. Among substrates of Bcr- Abl TK, the phosphorylation status of CrkL has been shown to be informative for the prediction of treatment outcome. White et al. has demonstrated that the relationship between the in vitro concentration of IM needed to reduce the level of phosphorylated CrkL (pCrkL) and the achievement of MMR, i.e, the lower IC50 was correlated with the higher MMR [60]. Moreover, patients who had greater than 50% in vivo inhibition of pCrkL within the first month of treatment had increased rates of MMR at 12 and 24 months [61]. IM plasma trough concentration also significantly associates with treatment outcome, namely, a minimum of trough concentration of 1002 ng/ml was associated with and optimal response [62]. The activity of drug influx pump, hOCT1, at diagnosis has been also shown to be associated with the treatment outcome of CML, when treated with IM, but not SGIs. According to two clinical trials, TIDEL (Trial of Imatinib with Dose Escalation) and TOPS (Tyrosine Kinase Inhibitor Optimization and Selectivity), it was found that MMR rates of patients with high hOCT1 activity and patients with low hOCT1 acitivty by 5 year treatment with IM were 89% and 55%, respectively [18].
4. Impact of load of Abl mutated clones on effects of second-line sgis: Because the in vitro kinase inhibitory profiles of SGIs differ, it is essential to screen Abl KD mutation to determine the optimal choice of SGIs as second-line treatment for IM-resistant patients [63,64]. An Australian group reported interesting results about this issue at the Annual Meeting of the American Society of Hematology in 2010, showing that the load of SGI-resistant Abl KD mutated clones at baseline affects the response to SGIs. CCyR was infrequent in patients with an SGI-resistant Abl mutated clone identifiable by direct sequencing (DS) (sensitivity: 10-20%), while its frequency was 16% for patients whose clone was identifiable by high throughput chip-based mass spectrometry (Mass Spec) (sensitivity: 0.05-0.5%) after 18 months of SGI treatment. In addition, expansion of Mass Spec-detectable minor clones co-occurring with SGI-resistant Abl KD mutation was detected in 84% of patients during SGI treatment. In contrast, the CCyR rate was 42% for patients with an SGI-nonresistant Abl KD mutated clone regardless of the load of mutated clones.
The next question is whether the presence of a clone with an SGI-non-resistant Abl mutation at baseline affects the course of treatment with SGIs. In the Australian study mentioned above, it was reported that even SGI-non-resistant clones increased in 12% of patients during treatment with SGIs. Moreover, even in cases with SGI-non-resistant Abl KD mutation(s) at baseline, SGI-resistant Abl KD mutated clones emerged in 34% of patients with a single SGIsensitive Abl KD mutation and in 67% of patients with two or more different clones with SGI-sensitive Abl KD mutations. As a result, the CCyR rate for patients with a single clone was 67% and was reduced to 14% for patients with two or more different clones. At the same meeting, groups from MD Anderson Cancer Center reported further impairment of PFS and OS in patients with multiple Abl KD mutations. From the view of clonal evolution, the presence of Abl KD mutated clones indicates not only sensitivity to SGI but also to what degree leukemic cells have already acquired genetic instability at the time of investigation. Putting these findings together indicates that the identification of Abl KD mutation is indispensable, but does not satisfactorily predict the effects of SGIs. Regardless of in vitro sensitivity to SGIs, the presence of Abl KD mutations implies a genetic instability which may impair the effect of SGIs. Close monitoring of the effects of SGIs is thus required under such circumstances.
5. Factors other than Abl KD mutation which impact the effects of sgis as second-line treatment: The in vitro kinase inhibitory profile cannot fully determine the clinical outcome of second-line SGI treatment. No effects of SGIs can of course be expected in the presence of T315I mutation, while the CyR rates attainable in the clinical setting with second-line SGIs are not significantly different for patients with SGI-moderately sensitive and patients with SGIsensitive Abl KD mutation, indicating that factors other than Abl KD mutations also play a role in determining clinical outcomes [37,65,66]. Accumulating evidence has made it clear that poor risk factors for second-line SGIs include non-MCyR with IM at 12 months, more than 4% basophils in peripheral leukocytes at the switch to SGIs, hemoglobin less than 12g/dL, the presence of additional chromosomal abnormality, poor Sokal score, or neutropenia during IM treatment [67,68]. All these poor risk factors show a positive association with clonal evolution and disease progression in CML, and the evidence presented here suggests the importance of disease control with TKIs whenever CML clones prove to be addictive to Bcr-Abl TK.
6. Monitoring of effect of second-line sgis during treatment: The long-term effect of second-line SGI is also predictable by monitoring the effect of ongoing SGI treatment. Because the effects of SGIs as second-line treatment are limited, it is essential to determine whether the ongoing SGI treatment is appropriate. Several findings have demonstrated the importance of treatment response after 3 months of SGI therapy. Hughes et al. demonstrated that the monitoring of bcr-abl copy numbers can help predict possible attainment of CCyR and MMR by using second-line SGIs. In this study, the rates of CCyR and MMR for patients who showed reduction of bcr-abl copy numbers to ≦1%, 1~10% and >10% after 3 months of SGI treatment were 91% and 86%, 56% and 55%, and 11% and 4%, respectively, after 24 months of SGI treatment [69]. Another study showed that the attainment of at least minor CyR after 3 months of SGI treatment was associated with later attainment of MCyR and better OS and EFS [70]. These findings may prove useful for deciding whether to switch treatment to another SGI or even to allogeneic HSCT.
7. The fight against T315I: At the timing of writing, no FDA-approved Abl-specific TKIs had been proven effective for leukemic cells with T315I Abl KD mutation, so that allogeneic HSCT remains one of the most effective strategies against T315I [71,72]. However, several new agents designed to fight T315I are under development. One of the most promising of these agents is ponatinib (formerly known as AP24534). Ponatinib is similar to IM and nilotinib in terms of its limited binding ability to the inactive conformation of Bcr-Abl but was developed as a scaffold that did not need to create a hydrogen bond with T315, and would therefore be effective for inhibiting Abl KD with T315I mutation. Another difference from IM and nilotinib is that ponatinib is active against P-loop mutations, such as E255K, Y253F, or G250, and, while it is effective against Src, Lyn, c-KIT, VEGFR2, PDGF-R and FGFR1, it is most effective against wild type Abl among a series of target kinases [33]. Cortes et al. reported the results of a finalized Phase I study of the efficacy of ponatinib for patients with advanced Philadelphia-positive leukemias, including 64 CML patients refractory to one to three TKIs and 10 with Philadelphia-positive acute lymphoblastic leukemia (ALL) [7]. Of the 38 CML-CP patients, 66% attained MCyR and 53% CCyR, of the 12 CML-CP patients with T315I, all attained CHR and MCyR and 89% CCyR, and MMR was attained by 42% of CML-CP patients, including 7 with T315I. Finally, adverse events were tolerable in most cases. A phase II study of ponatinib (Ponatinib Ph+ acute lymphoblastic leukemia (ALL) and CML Evaluation or PACE) is currently under way. Moreover, several other agents, such as DCC-2036, a switch pocket inhibitor [73], or danusertib (PHA-739358), XL-228 and AT-9283, aurora kinase inhibitors, are being developed as potent therapeutics for IM/SGI-refractory CML, including patients with T315I Abl KD mutation [7,74]. Danusertib is a pan Aurora and Abl inhibitor and has been evaluated in a Phase I study for 12 advanced phase CML and 11 Ph1-positive ALL patients, including 15 with T315I mutation. Danusertib induced hematologic response in five and cytogenetic (any) response in three patients in those series. XL- 228, a multi-kinase inhibitor for Aurora A, Abl, IFG-1R, Src and Lyn, has been also evaluated in Phase I study for 27 patients with CML and Ph1-positive ALL, 10 of which had T315I mutation. In this trial, objective responses have been reported in some patients, including patients with T315I. The effect of the pan-Aurora, Abl, FLT3 and JAK2 inhibitor AT9283 has been also reported against CML patients who were refractory to IM and dasatinib [74]. The final results of those trials are awaited.
8. Complete cure with tkis? A recent study by a French group of patients who maintained complete MR (CMR) for more than two years by IM treatment has shown the possibility of cure by IM treatment for some patients. Even in case of relapse following the cessation of IM treatment, the reintroduction of IM was effective for those patients. A higher probability of sustained CMR following the cessation of IM treatment was observed for patients with a low Sokal risk score and longer IM treatment (≧ 50 months) [75]. These results suggest two important hypotheses. One is that low-risk, perhaps early phase, CML-CP, which is highly addictive to Bcr-Abl TK, may be curable with intense and long-term TKI treatment. The other is that even for non-cured patients, the long-term and deep suppression of leukemic clones may reduce the risk of clonal evolution which may cause the loss of ddiction to Bcr-Abl TK activity. The faster and the deeper curative effects of SGIs on newly diagnosed CML-CP clearly point to the urgent need to investigate the potential potency of SGIs for a complete cure of CML.
Future perspective of pharmacologic interventions for cure of CML
Finally, we address the possible new treatment strategies by the combination of TKI and other anti-leukemic agents for cure of CML. Rationally, a strategy which blocks Bcr-Abl signaling as the main pathway using TKIs and the simultaneous blockade of collateral signaling pathway by partner agents may provide the best chance for disease elimination. Especially, combination strategies which completely eliminate leukemic stem cells (LSCs) may be desirable, because CML LCSs are insensitive to IM and persist as the cause of relapse. Considering this aspect, the combination of agents those potentially target molecules essential for LSC survival and maintenance may be of interest. Homoharringtonine (HHT), a Chinese evergreenderived alkaloid cephalotaxine, simultaneously inhibits Mcl-1 and β-catenin, those are crucial for the survival of LSCs, and it indeed effectively kills LSCs in preclinical model [77,78]. According to the result of the trial for the combination of IM, G-CSF and HHT against advanced phase IM-resistant CML, this combinatory strategy was most likely feasible [76]. It will be also interesting to look at the combinatory effects of IM and HHT for CML-CP. Also, the combination of IM and decitabine, a DNA methyltransferase inhibitor, has been shown to be well-tolerable and active against advanced phase CML [79]. The effects of DNA methyl transferase inhibitors on the gene expression modification in LSCs are expected to enhance the clinical effect of IM in CML-CP. Farnesyl transferase inhibitor (FTI) which potentially inactivates Ras-related proteins and MEK inhibitor have been shown to be active against CML LCSs [80], and the combination of lonafartinib, a member of FTIs, and IM has been shown to be well-tolerated against CML patients who failed IM monotherapy in Phase I trial [81]. It would be of interest to investigate the effect of this combination against CML-CP as the first-line setting.
In this article, we reviewed the current knowledge regarding TKI treatment for CML. The success of TKI treatment for CML constitutes the paradigm of molecular targeted therapy for malignant diseases and has shown the way towards the development of new treatment for cancers as well as how problems involving new therapeutics can be tackled. Making further progress on the basis of the new and updated information is becoming increasingly important for both clinicians and researchers.