Journal of Physical Chemistry & Biophysics
Open Access
ISSN: 2161-0398
ISSN: 2161-0398
Research Article - (2014) Volume 4, Issue 2
This study reports the development of a novel formulation of a polymeric nanoparticls (NPs) with the drug Promazine hydrochloride (PRO), a hydrophobic molecule, dispersed in biodegradable polymeric matrix of poly (DLlactide- co-glycolide) (PLGA) by using emulsion-solvent evaporation method at the temperature T = 298.15 K. Spherical NPs with controlled size were designed. PRO was capsulated into nanoparticles with theoretical drug loading (TDL) varying from 10 to 30% (w/w). The effects of TDL, of poly(vinyl alcohol) (PVA) concentration, of PLGA concentration in organic phase, the effect of power of sonication and of pH of aqueous phase were studied. After lyophilization of PRO-loaded nanoparticles, the average size, Zeta potential, and polydispersity index at TDL 30% were 350 ± 22 nm, −18.7 ± 2.0 mV, and 0.18 ± 0.04, respectively (at TDL 30%, PLGA content 1.3 % w/v and pH = 9). The maximum drug encapsulation efficiency and drug loading capacity were 32.74 ± 0.54% (w/w) and 19.13 ± 0.38 %, respectively (at TDL 30%, PLGA content 1.3% w/v and pH = 9). Scanning electron microscopy studies showed spherical and smoth shape of drug-loaded nanoparticles. Solid lyophilized NPs were evaluated for in vitro release in phosphate–buffered saline (pH = 7.4) by using dialysis bags. Parameters for the release process showed that both the initial PLGA content and energy of sonication have no significant influence on PRO release from NPs.
<Keywords: Experimental nanomedicine; PLGA; Promazine hydrochloride; Controlled drug delivery; Physicochemical properties of nanoparticles
Many well known and recently developed drugs encounter delivery issues due to their poor aqueous solubility. Phenothiazine derivatives have been shown to be highly effective as constituents of neuroleptics revealing antipsychotic properties [1,2]. They belong to a big group of tricyclic aromatic compounds. They easy react with halide and organic complexes with metals and form well-defined ion-associated complexes [3]. Due to their pharmacological properties and potent toxicity, they have become group of drugs with increasing interest in clinical study. Promazine and triflupromazine were used with satisfactory results in breast cancer therapy in combination with doxorubicin [4]. Results suggested that phenothiazine derivatives were highly toxic for both cell lines, with triflupromazine showing the highet effects.
Halogenation of drugs is commonly used to enhance membrane binding and permeation. Thus in this work, the one of hydrochlorides of phenothiazine derivatives, the Promazine hydrochloride (PRO) was investigated. It is continuation of our previous work on solubility of PRO in alcohols as well as on developing new pKa values of PRO [5] and of similar drugs [6]. The values of pKa and intrinsic solubility, S0 (53.5 μmol ) for PRO were published many years ago, however without the information about pH [7]. The interfacial membrane partitioning and permeation of PRO was developed and the lipid-water partition coefficient was calculated and measured by the titration calorimetry [8].
In recent years, significant effort has been devoted to develop nanotechnology for drug delivery. Nanotechnology focuses on formulating therapeutic agents in biocompatible nanocomposites such as nanoparticles, nano-capsules or micellar systems [9,10]. Polymeric nanoparticls (NPs) have a polymeric shell and an inner core. The active drug is usually dissolved in the core, but may also be packed in the surface [9]. NPs have been extensively investigated in drug delivery systems for drug targeting because their particle size ranging from 10 to 800 nm and is acceptable for intravenous injection [11-16]. NPs have been extensively studied for lipophylic drug delivery in recent years and appear to be the best form of oral drug delivery in the future. The proper drug delivery system should be not only biocompatible and biodegradable, but also poorly immunogenic, able to load the drug efficiently and make no changes in activity of drug. This is extremely important for chemotherapeutic drugs. NPs with drugs are being increasingly used to improve drug efficacy, specificity, tolerability and therapeutic index [17]. Up to date, researchers are still looking for an ideal nano-carrier method for the controlled delivery of poorly watersoluble drugs [14-16]. During the last decade, a number of different polymers have been investigated for formulating biodegradable NPs. However, the most popular was biodegradable poly(L-lactic-acid) in copolymer with glycolic acid giving poly(DL-lactide-co-glycolide) (PLGA) [12,14-16,18-20]. The lactide/glycolide particles are not toxic to body and are eliminated from the body by the citric acid cycle as lactic and glycolic acids [9]. The PLGA polymer is hydrophobic and soluble in chloroform and dichloromethane and mainly form nanospheres with drugs, delivered to the cells [16]. The process strongly depend on pH of the solution. The effect of pH on the solubility of drug of ionisable compounds is well known and was recently summarized in an excellent review about solubility of drugs [21].
The drug release from NPs depends on many processes: drug physicochemical properties (including solubility in water), drug-PLGA interactions, loading of NPs, diffusion of drug from the core to the surface, diffusion through the polymer, which depends on polymer degradation and pores and channels concentration in the polymer. However, the release of drug as a function of time profiles is in many times to be diffusion controlled and described by various models based on Fick’s law of diffusion [22]. The information obtained from release profiles allow new predictive methods for the technological new formulations. Three methods for the release of drug is usually used: continuous flow method, separation technique, and membrane diffusion technique [23].
The release of drug encapsulated micro particles may be diffusion controlled, dependent on polymer degradation, or may occur by a combination of drug diffusion and polymer degradation.
The aim of the present study was to formulate a new PLGAbased drug delivery system with variables on size distribution of NPs including the PRO drug inside the core of polymer with a drug controlled delivery. PRO was chosen as a model drug for proposed emulsion-solvent evaporation method. The second objective of this work was to examine the effect of PRO release from NPs being mainly dependent on the amount and character of polymer, and loading of NPs. Thus the influence of different parameters controlling release profiles was investigated.
Chemicals and reagents
Poly (D,L-lactide-co-glycolide acid) (PLGA), as a copolymer ratio of D,L-lactide to glycolide of 50:50 (Mw 54,000-69,000 g/mol) was purchased from Sigma-Aldrich, Germany. Promazine hydrochloride (PRO) was purchased from Sigma-Aldrich, Finland (CAS number 53- 60-1). The poly vinyl alcohol (PVA) (95% hydrolysis degree and Mw 95,000 g/mol), supplied from Across-Organics, USA (CAS number 9002-89-5) was used as the surfactant in the emulsification process. Visking dialysis bags with pore size of 12kDa were obtained from Sigma-Aldrich, Germany. All chemicals: dichloromethane (CAS number 75-09-2, 0.999 mass fraction) acetonitrile (CAS number 75-05- 8, 0.99 mass fraction) and dipotassium hydrogen phosphate, K2HPO4 (CAS number 7758-11-4, 0.999 mass fraction) were purchased from Sigma-Aldrich, Germany. The phosphate buffer pH = 9.0 was prepared of potassium hydroxide, KOH (CAS number 1310-58-3, 0.995 mass fraction purity), potassium dihydrogen phosphate, K2HPO4 (CAS number 7758-11-4, 0.995 mass fraction purity), both purchased from POCH, Poland and phosphate buffer pH = 7.4 was prepared of the mixture of (KH2PO4 + K2HPO4).
Solvents were analytical grade and used without further purification. Twice distilled and degassed water deionised and filtered with Milipore Elix 3 was used for the aqueous solutions of drug.
Preparation of nanoparticles
Drug-loaded PLGA NPs were prepared by an emulsion-solvent evaporation technique using PVA (0.5% w/v) as surfactant. In brief, the drug and PLGA was dissolved in the 3 mL of dichloromethane to obtain concentration of 10%, 20% and 30% w/w of drug to PLGA in the solution. This organic phase (3 mL) was added drop wise into aqueous phase (10 mL, containing 0.5% w/v PVA). Aqueous phase was buffered to pH = 9.0 with the phosphate buffer, in order to minimize drug loss. The mixture was homogenized for 3 min with the help of probe sonicator set at level 30 % of power 70W (HD 2070, Bandelin Sonopuls, Germany). The organic phase was evaporated during 15 min using a rotative evaporator under partial vacuum. The NPs were isolated by centrifugation (6,000 rpm, 20 min, Hitachi) and washed twice with double-distilled water. The NPs were freshly used or lyophilized for investigation.
Nanoparticles characterization
The nanoparticles size and zeta potential were estimated on the zetasizer Nano (Malvern Instruments, Malvern, United Kingdom). For the measurement, 0.5 mL of freshly prepared and purified suspension of NPs was diluted in 5 mL of distilled water and the mixture was sonicated during 2 min. The analysis was carried out at a scattering angle of 90º and at a temperature of T = 298.15 K. The Z-average diameter, zeta potential and polydispersity index were calculated using Malvern software. For statistical analysis all samples were measured in triplicate and the average values and standard deviation of the measurements were calculated.
The surface morphology of NPs was examined by scanning electron microscopy (SEM, FEI Quanta 200, USA). The samples of dray, solid, lyophilized NPs were placed on metallic surface and next were drayed under vacuum. Observations were obtained at 20 kV.
Assay of encapsulation efficiency
The concentration of drug encapsulated in nanoparticles was determined spectrophotometrically using the Spectrometer UV-vis Lambda 25 (PerkinElmer Life and Analytical Sciences, Shelton, USA) at chosen wavelength, λ = 255 nm. The reference cuvette was filled with the mixture of acetonitrile and water, 1:4 v/v. The linear calibration curve for PRO was obtained in the same mixture of acetonitrile with water (1:4 v/v) in the range from 3 μg/mL to 15 μg/mL (R2 = 0.998). The overall experimental uncertainty for the temperature was estimated to be ± 0.05 K. Photometric accuracy (NIST 930D Filter 1A) obtainable with UV-Vis Spectrophotometer is ± 0.001 A and repeatability ≤ 0.001 A. The uncertainty in composition was 1 · 10-6 mol · dm-3. Many series of UV spectra were recorded at T = 298.15 K, and for each sample the obtained peaks were analyzed. The calibration curve for the chosen wavelength λ = 255 nm is shown in Figure 1 and the absorbance vs. wavelength for chosen system in Figure 2.

Figure 1: Linear calibration curve for PRO in mixture of acetonitrile and water (1 : 4 v/v).

Figure 2: Dependence of absorbance on the wavelength for PRO at T = 298.15 K for chosen mixture.
The indirect method was carried out by measuring the amount of PRO not encapsulated in PLGA NPs. Freshly formulated NPs solution was centrifuged at 6000 rpm/min during 20 min to separate solid NPs. Next, the free drug in the supernatant was essayed, assuming that drug not present in the supernatant was incorporated in PLGA NPs [24,25]. The encapsulation efficiency (EE) was calculated as follow:
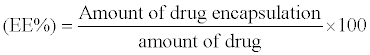 (1)
(1)
The drug loading determined as follow:
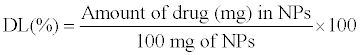 (2)
(2)
Drug release studies
Lyophilized NPs were evaluated for in vitro release in phosphate– buffered saline (pH = 7.4) by using dialysis bags. Firstly NPs were resuspended and dispersed by sonication in 1 mL of buffer and then poured in dialysis bags. The dialysis bags were soaked in distilled and deionised water for 12 h before use. The bags then were placed into the bottle containing 60 mL of release medium (buffer, pH = 7.4) at temperature T = 310.15 K under magnetic stirring at 100 rpm. At each selected time point, aliquots of 1 mL were withdrawn and analyzed spectrophotometrically 2.4 to determine the amount of released PRO.
Preparation and characterization of PLGA NPs
PRO–loaded NPs were prepared by emulsion evaporation technique in varied processes, dependent on many parameters in order to obtain optimal formulations conditions: theoretical drug loading of PRO, PVA concentration in the formulation, PLGA content in the formulation, the power of sonication, and the pH of the aqueous phase. Only one or two parameters were changed in each series of experiments.
Theoretical drug loading of PRO (TDL)
The actual drug loading of NPs depends on the TDL. Three different TDLs of 10%, 20% and 30% w/w were selected to oversee the effects of TDL on actual drug loading - drug entrapment and the size of NPs.
An increase in the content of PRO has a remarkable effect on the practical drug loading, drug encapsulation and the particle size (Table 1 and Figure 3). It can be observed that an increase of the TDL form 10 to 30% increases the size of NPs gradually from 362.3 nm to 393.2 nm. This is the result of the increasing of the drug content in NPs. It is shown in Figure 3 that drug loading increases systematically with an increase of TDL for pH=9. However, the encapsulation efficiency is changing irregularly with an increase of TDL. Not the first time, an increase of the TDL results in an increase of the viscosity of dispersed phase, which increases the size of the NPs [12,26]. The particles formulated by the emulsion evaporation method have a nanoscale and quite low polydispersity index, PDI measured by the zetasizer Nano. The low values of PDI indicate that nanoparticles have a narrow particle size distribution. The zeta potential of nanoparticles is an important aspect to determine the surface charge of nanoparticles. The PLGA NPs loaded with PRO have a negative charge (-15.2 ± 0.74 to -18.7 ± 2.00) as is shown in Table 2, which could be related to the presence of the end of carboxyl groups on the surface of NPs [27].

Figure 3: The effect of theoretical drug loading on ( □) encapsulation efficiency and (■) drug loading (pH = 9).
| Name of compound | Structural formula | Mw/(g∙mol-1) |
|---|---|---|
| Promazine hydrochloride (PRO) | 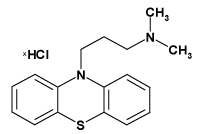 |
320.90 |
| Poly(DL-lactide-co-glycolide) (PLGA: 50:50) |
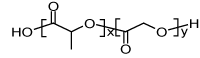 |
54.000-69.000 |
Table 1: Investigated compounds: name, abbreviation, structure, and molar mass.
| Theoretical drug loading (% w/w) | Particle size (nm ) | PDI | Zeta potential (mV) |
|---|---|---|---|
| 10 | 362.3 ± 12.0 | 0.16 ± 0.010 | - 18.4 ± 1.56 |
| 20 | 374.1 ± 18.8 | 0.22 ± 0.055 | - 15.2 ± 0.74 |
| 30 | 393.2 ± 14.91 | 0.19 ± 0.007 | -18.7 ± 2.00 |
Table 2: Effect of theoretical drug loading on particle size, polydispersity (PDI), the Zeta potential, encapsulation efficiency and drug loading (DL) at PVA = 0.5 (% w/v), PLGA = 0.8 (% w/v), pH = 9).
In order to reduce the drug wastage, the encapsulation efficiency must be high. As it is shown in Figure 3, the highest encapsulation efficiency was observed at 10 TDL (34.23 ± 1.80 w/w) with final TDL of (4.99 ± 0.35 w/w). Furthermore, an increase in the concentration of drug in organic phase leads to an increase in actual drug loading (15.23 ± 4.90).
Effect of PVA concentration
Previous attempts to encapsulate PRO in PLGA NPs resulted in low encapsulation efficiency and poor stability. In this study, PVA was used additionally as a surfactant. The mean particle size, encapsulation efficiency and the drug loading of PRO in PLGA NPs as a function as the various concentrations of PVA in aqueous phase are listed in Table 3 and are shown in Figure 4. As observed from the measurements, the decrease of PVA concentration causes the reduction of NPs size (the size of NPs obtained at 0.3% w/v concentration of PVA was 291.6 ± 25.6 nm). The increase of concentration of PVA to 0.7 % w/v leads to formulation a bigger NPs size (317.9 ± 30.05 nm). Furthermore, the PVA concentration has an significant role in emulsification process, the stabilization of emulsion and the protection of the droplets from coalescence [28,29]. The organic phase easily disperses in aqueous phase at low PVA concentration, which results in low viscous solution and consequently a smaller NPs sizes. In higher concentration of PVA, the viscosity of aqueous phase increases and leads to associate the droplets, which results in the bigger particles obtained. Additionally, Figure 4 depicts that the higher PVA concentration can stabilizing the effects of surfactant in the solution and reveals higher encapsulation efficiency (at PVA 0.7 % w/v, the encapsulation efficiency is 61.62 ± 2.42) and DL is 36.55 ± 1.60 (at PVA 0.7 % w/v). Thus, the increase of PVA concentration results in significant multiply the encapsulated efficiency and drug loading. At high concentration of PVA the encapsulation efficiency was three times larger than that for PVA 0.3 % w/v. Increasing of viscosity can avoid diffusion of drug into the aqueous phase and results in increase of the drug entrapment into NPs. The changes in polydispersity are not large (Table 3).

Figure 4: The effect of the PVA concentration in aqueous phase on the ( □ ) encapsulation efficiency and (■) drug loading (DL) (pH = 9).
| PVA concentration (% w/v) | Particle size (nm ) | PDI |
|---|---|---|
| 0.3 | 291.6 ±25.57 | 0.145±0.030 |
| 0.5 | 349.3 ± 21.82 | 0.176 ± 0.037 |
| 0.7 | 317.9 ±30.05 | 0.156 ±0.072 |
Table 3: Effect of the PVA concentration in aqueous phase on the particles size and polydispersity (PDI) at TDL 30 (% w/w), PLGA = 1.3 (% w/v), pH = 9.
Effect of PLGA concentration
The effect of PLGA concentration on mean particle size, polydispersity, drug encapsulation and drug loading was also studied. PLGA concentration varied between 0.8 and 1.6 (w/v), which changes the particle size between 393.2 ± 14.91 and 344.5 ± 14.3 at TDL 30 (% w/w), PVA concentration 0.5 (% w/v) and pH = 9. The results are listed in Table 4. Figure 5 provide the information about the influence of PLGA concentration on an encapsulation efficiency and DL at the TDL 30 (% w/w). It can be seen that both an encapsulation efficiency and DL increase when the PLGA concentration increases. From our other experiments it was obtained that the PLGA content causes an increase of the particle size from (0.8% w/v; 362.3 ± 12.02 nm) to the 1.3 (% w/v; 370.0 ± 9.95 nm) at the TDL 20 (%w/w). This result was similar to the conclusions obtained by other authors [12,30]. Unfortunately, at the TDL 30 (%w/w) the opposite dependency on particle size is observed (0.8 w/v; 393.2 ± 14.91 nm) has changed to the (1.3% w/v; 344.5 ± 14.30 nm) (Table 4). This is quite non-understandable because usually an increase on the concentration of PLGA leads to an increase of NPs size due to the increase of viscosity of the organic phase. In more viscous phase a poorer dispersability of the organic to aqueous phase should be expected. In our experiment, it is no doubt the influence of TDL. Polydispersity is low depended on PLGA concentration and it is on the level of 0.17 to 0.19. Most of our experiments were performed at the TDL 30 (% w/w), thus the influence of PLGA on encapsulation efficiency and the DL increases. This is related to increase of viscosity of organic phase. Increasing viscosity of organic phase can strengthen the drug resistance to diffuse into aqueous phase and thus increased the drugs encapsulation into NPs.

Figure 5: Effect of PLGA concentration on (□) encapsulation efficiency and (■) drug loading (DL) at the TDL 30 (% w/w) (pH = 9).
| PLGA concentration (% w/v) | Particle size (nm ) | PDI |
|---|---|---|
| 0.8 | 393.2 ± 14.91 | 0.192 ± 0,007 |
| 1.3 | 349.3 ± 21.82 | 0.176 ± 0.037 |
| 1.6 | 344.5 ± 14.30 | 0.188 ± 0.004 |
Table 4: Effect of PLGA concentration on particles size and polydispersity (PDI) at the TDL 30 (% w/w), PVA = 0.5 (% w/v), pH = 9.
The SEM experiment for chosen samples is shown in Figures 6 and 7. SEM images of PRO-loaded PLGA prepared with PVA concentration 0.5 (% w/v), PLGA concentration of 0.8 (% w/v) and pH = 9 show spherical shape and absence of both agglomeration and amorphous polymer (Figure 6). However, NPs prepared with PLGA concentration of 1.3 (% w/v) (the rest of parameters were kept constant) presents spherical shape as before, the lower particle size (at the TDL 30 % w/w) and presents some agglomerates (Figure 7). This may be explained by the greater probability of the association of molecules in a more concentrated solution.

Figure 6: SEM images of PRO-loaded PLGA prepared with PLGA concentration 0.8 (% w/v).

Figure 7: SEM images of PRO-loaded PLGA prepared with PLGA concentration 1.3 (% w/v).
Effect of sonication and of pH of aqueous phase
In order to obtain lower particle size in emulsified system, the increase of energy of sonication is a fundamental step. The effect of power of sonication and of pH of the aqueous phase on particle size, polydispersity, encapsulation efficiency and DL at the TDL 30 (% w/w) is shown in Table 5. As was expected, an increase of power of sonication from 30% to 60% of power 70W at constant pH = 9 decreases the particle size from 349.3 ± 21.82 nm to 327.5 ± 15.42 nm and slightly decreases PDI. The encapsulation efficiency and the DL increases. Decreasing of pH from 9 to 7.4 significantly decreases the particle size to 249.6 ± 9.79 nm but also decreases all of the rest parameters: polydispersity, encapsulation efficiency and DL. A comparison of different results at the temperature T = 298.15 K shows that the pH = 9 is better than pH = 7.4 because of large decrease of drug loading for lower pH. It can be concluded from the obtained results that higher energy released in the emulsification process leads to a better dispersion of polymeric organic phase as nanoparticles of smaller size and higher DL. This effect was observed also by other authors [12,31].
| Power of sonification | pH aqueous phase | Particle size (nm ) | PDI | Encapsulation efficiency | DL (%) |
|---|---|---|---|---|---|
| 30 | 7.4 | 249.6 ± 9.79 | 0.088 ± 0.022 | 7.12 ± 1.44 | 4.22 ± 0.83 |
| 30 | 9 | 349.3 ± 21.82 | 0.176 ± 0.037 | 32.74 ± 0.512 | 19.13 ± 0.38 |
| 45 | 9 | 333.0 ± 45.80 | 0.136 ± 0.026 | 41.09 ± 8.00 | 25.29± 4.65 |
| 60 | 9 | 327.5 ±15.42 | 0.137 ± 0.017 | 52.82 ± 6.14 | 31.74 ± 3.63 |
Table 5: Effect of power of sonication and of pH of the aqueous phase on particle size, polydispersity (PDI), encapsulation efficiency and drug loading (DL) at the TDL 30 (% w/w), PVA = 0.5 (% w/v), PLGA = 1.3 (% w/v).
The release experiment
The in vitro drug release profile from NPs at temperature T = 310.15 K in phosphate–buffered saline, pH 7.4 (the body pH) for the same amount of PRO (1.986 mg, 2.006 mg and 1.916 mg), for different concentrations of PLGA (% w/v) 0.8, 1.3 and 1.6 and for different sonication set at 30%, 45% and 60% (of 70W) is shown in Figure 8 a and b, respectively. The PRO released was studied as a function of time during 48h. There is no significant difference in release for different amount of PLGA and sonication time. About 75 w/w % of PRO was released already from NPs after 4h for sonication 30% and contamination of PLGA 1.3 or 1.6. The character of dependences is the same. Minimally faster release was observed for 0.8 of PLGA (% w/v) in the sample.

Figure 8: The in vitro drug release profile from NPs at T = 310.15 K in phosphate–buffered saline, pH 7.4 for the same amount of drug: a) results for different concentration of PLGA (% w/v): (▲) 0.8 (power of sonication set at 30 % of 70 W); (♦ ) 1.3 (power of sonication set at 30 % of 70 W); (■) 1.6 (power of sonication set at 30% of 70 W); b) results for the different power of sonication: (♦ ) 30%; (■) 45%; (▲) 60% at concentration of PLGA, 1.3 (% w/v).
The results discussed above for PRO release from NPs show that the inclusion of PRO at pH = 9 leads to the degradation of the PLGA during processing. The use of buffer systems has strong influence on the solubility of the drug in water [21] and on the degradation of polymer system [14].
We employed the emulsion-solvent evaporation method allowed the preparation the drug-loaded nanoparticles of promazine hydrochloride in biodegradable PLGA polymer under different conditions. The fast solvent diffusion provided to the small, submicron and spherical nanoparticles, which mean size, encapsulation efficiency and drug loading, depended on theoretical drug loading, PVA concentration, PLGA concentration, energy of sonication process and pH of the aqueous phase. In this work, the proces parameters of novel nano-formulation of promazine hydrochloride were optimized to produce high encapsulation efficiency. We combined different factors to obtain the smaller particles with the highest drug loading and the smallest polydispersity. The conclusion can be made that the increase of pH of the aqueous phase from 7.4 to 9.0 has a significant effect and increases the encapsulation efficiency and drug loading (unfortunately increases also the particle size). Moreover, by increasing in concentration of PVA, the higher encapsulation efficiency of 61.62% and the smaller NPs size of around 317.9 nm were obtained. It can be suggested for the future work to use larger concentration of PVA, for example 0.7 % w/v. In this work, most of the measurements were made for TDL = 30%, PVA concentration = 0.5 (% w/v), PLGA concentration = 1.3 (% w/v), sonication power, 30% of 70 W and pH = 9.
After tree different measurements, it is obvious that the drug is inside the polymer cover: i) time of release is few hours not few minutes, which means that drug is inside the nanoparticle; ii) Zeta potential is measured for the polymer surface no for the drug; iii) SEM images of PRO-loaded PLGA show sharp border of nanoparticles rings -when the drug is on the surface of nanoparticles there is usually foggy picture around.
In conclusion, the preparative variables can be exploited in order to obtain optimal condition for formulation PLGA nanoparticles loaded with PRO. This study showed also that there is no significant difference in release kinetics of PRO for different amount of PLGA and sonication time. The obtained PLGA nanoparticles have potential to produce sustained release of promazine hydrochloride.
Funding for this research was provided by the Warsaw University of Technology.