Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2017) Volume 10, Issue 4
It is expected that we can predict the mechanism of folding of a protein from only its sequence if the information about the folding is encoded in the sequence. This is a long standing problem in molecular biophysics and molecular bioinformatics. In this study, we examine the folding of the lysozyme-like superfamily proteins that diverged from a common ancestor protein and show the common function and partially common 3D topology. We use methods based on inter-residue average distance statistics and evolutionary analysis to identify the location of the folding region from only their amino acid sequence. The properties of these sequences indicate that the general folding mechanisms are common among proteins in the same family. The difference in the folding mechanisms between the lysozymes in the animal kingdom and that in λ-phage are also suggested. We compare the hydrophobic packing observed in the actual 3D structures of the lysozymes with the results of the present study. We confirmed that the possible significant residues predicted in this study form hydrophobic packing in the 3D structures of the examined lysozymes. The sequence properties in the partially common 3D topology of these proteins imply that these parts are highly involved in the folding mechanisms in each family. We also find out that hydrophobic interactions of partially common 3D topology appeared in all families are conserved. These conserved hydrophobic contacts may be significant for forming the common topology; these contacts can be regarded as significant for the common function.
Keywords: Lysozyme; Folding; Evolutionary analysis; 3D topology
How a protein folds to its native structure is a long standing problem in molecular biophysics and molecular bioinformatics. It should be possible to predict the mechanism of folding of a protein from its amino acid sequence if all information of protein folding is encoded in its sequence. It is rather difficult to extract the information of folding of a protein from its sequence based on only standard bioinformatics techniques such as multiple alignments of homologous sequences.
It is well known that the 3D structures of proteins in lysozyme-like superfamily show wide variety but partially common 3D topology [1-3]. It has been also pointed out that the common 3D topology relates to their common function, that is, bacteriolysis [4]. How is the information on the partially common 3D topology of lysozyme-like superfamily proteins encoded in their sequence? This seems to be very fundamental but difficult question. In this study, the following lysozyme families are selected: C-type lysozyme family and its superfamily proteins I-, G-, and L-type lysozyme family. The 3D structure of a protein from V-type lysozyme also shows correspondence to their common 3D topology, but its detailed 3D structure seems to deviate from the common topology of other lysozymes. Therefore, we exclude V-type lysozyme from the present study.
Figure 1A-1D present the schematic drawings of the 3D structures of the representative members from C-, I-, G-, and L-type lysozymes, that is, hen egg white, Tapes japonica, goose, and λ-phage lysozymes. A secondary structure in the common topology with a same color expresses a structural commonality in all lysozymes. Proteins treated in this study exhibit about 30% sequence identities within a same family, but proteins from different families show about 10-20% identities. This fact suggests the difficulty in recognizing the similarity of partial 3D structure among these proteins only by means of sequence alignment. Do these partially similar 3D structures form via a common folding mechanism? It would be very interesting to answer this question.

Figure 1: The schematic drawings of 3D structures of (A) hen egg white lysozyme (HEWL), (B) Tapes japonica lysozyme (TjL), (C) goose lysozyme (GL), (D) λ-phage lysozyme (LaL). A cylinder and an arrow denote as α-helix and a β-strand, respectively. A secondary structure of the same color is structurally common in all lysozymes.
The folding mechanisms of two C-type lysozymes, that is, goat α-lactalbumin and canine milk lysozyme were investigated by Nakamura et al. by means of ϕ-value analyses at the Ca2+ binding sites and H/D exchange technique for molten globule states [5]. These proteins share 41% sequence identity and almost the same 3D structures. Their study has revealed the different distributions of peaks in the protection factors for these proteins and also the difference in non-local contact distributions in their native structures. Thus, their study demonstrated that the folding mechanisms of proteins with highly sequential homology may be different and that a lysozyme is sometimes affected not only by topology of its main chain but also side chains.
In the present study, we attempt to elucidate the folding mechanism of each protein from its amino acid sequence in the families mentioned above by means of techniques based on the inter-residue average distance statistics in combination with evolutionary analyses. We have been applying a contact map based on inter-residue average distance statistics (we refer it as average distance map analysis) and a contact frequency prediction technique also based on inter-residue average distance statistics for protein folding [6,7]. Examples of the applications of these techniques to extract the information of folding mechanisms are the following proteins: fatty acid binding proteins [8], globin-like fold proteins [9], IgG binding and albumin binding domains [7,10], immunoglobulin-like fold proteins [11], ferredoxin-like fold proteins [12], and β-trefoil fold proteins (to be published). We apply these techniques to the present problem. The results of the proteins from C-type and L-type lysozyme families are compared with those of the experimental studies and the validity of the present techniques is examined.
As the next step of this work, we analyse the sequences for the folding mechanisms of proteins in a lysozyme-like superfamily. We are interested in whether folding mechanisms in a family are conserved during evolution. In general, conservation and its extent of the folding mechanism of homologous proteins is a very interesting and significant topic [5]. Koga et al. pointed out the difficulty of this problem because of energetically unfavourable non-ideal features that arise in proteins from evolutionary selection for biological function or from neutral drift [13]. Furthermore, we compare the hydrophobic packing observed in the actual 3D structures of the lysozymes with the results of the present study. We also discuss the mechanisms of formations of the common 3D structure in proteins in the lysozyme-like superfamily.
Proteins treated in this study
We treat lysozyme-like superfamily proteins (according to SCOP classification [14]) distributed in the animal kingdom and in λ-phage. C-type lysozymes are widely distributed over vertebrate and invertebrate animals [15]. α-Lactalbumins are homologues of C-type lysozymes [16,17]. I-type lysozymes are only distributed in invertebrate animals [15]. G-type lysozymes show similar distribution to that of C-type lysozyme [15]. L-type lysozymes are distributed in temperate λ-phage and bacteria.
These lysozymes are considered to have diverged from a common ancestor protein [3,15]. Evolutionary phenomena such as gene duplication and gene loss produced sequential and structural diversity of lysozymes [15].
Search of homologues
In order to obtain homologues in each family of C-, I-, G-, and L-type lysozymes, homology searches were conducted by means of BLAST [18] using proteins in Table 1 as queries with the e-value of 0.01 to make sure to obtain evolutionary homologous sequences. In this study, we use the abbreviations HEWL, TjL, GL, and LaL for these query proteins, that is, hen egg white lysozyme, Tapes japonica lysozymes, goose lysozyme, and λ-phage lysozyme, respectively.
| Species of Protein Families | ||||
|---|---|---|---|---|
| C-type | I-type | G-type | L-type | |
| Protein name of queries | HEWL1 | TjL2 | GL3 | LaL4 |
| Uniprot ID of each query | P00698 | Q8IU26 | P00718 | P03706 |
| PDB ID of each query | 2VB1 | 2DQA | 153L | 1AM7 |
| Objective database | Swissprot | UniprotKB | UniprotKB | UniprotKB |
| 1Hen egg white lysozyme, 2Tapes japonica lysozyme, 3Goose lysozyme, 4l-phage lysozyme | ||||
Table 1: The query proteins of each family for BLAST search.
For the search of homologues of the query protein of C-type lysozymes, Swiss-Prot was used as a database because this database is considered to be well trusted because of its high quality annotation [19]. For the rest of families, UniprotKB was employed as a database [20] because Swiss-Prot includes only a few sequences for these families. We were careful to pick candidates of homologues by checking protein existence (PE) numbers [21]. We excluded sequences with length less than 85% of that of the query sequence, with more than 90% of sequence identity, and with a gap of more than five residues within a secondary structure from the searched sequences. (Uniprot IDs of proteins in this work are summarized in Table S1 of Supplementary Information 1).
Analyses with inter-residue average distance statistics
Contact map analysis based on inter-residue average distance statistics: Details to construct a contact map based on inter-residue average distance statistics are described in Ichimaru and Kikuchi; Kikuchi et al. [8,22]. Here we give a brief survey to construct a map from the inter-residue average distance statistics (average distance map, ADM). The detailed procedure to construct and analyse ADM is given in Supplementary Information 2. The inter-residue average distances and standard deviations were calculated using known 3D structures considering the amino acid types and sequence separation. The separation of two residues along a sequence is taken into account as follows; M=1 when 1 ≤ k ≤ 8, M=2 when 9 ≤ k ≤ 20, M=3 when 21 ≤ k ≤ 30, M=4 when 31 ≤ k ≤ 40, and so on, where k=|i-j| and M is called “range”. The average distances of all pairs of amino acid types were calculated in each range.
If the average distance of a pair of amino acids in a range M is less than a threshold value determined in advance, a plot is made on a map (the threshold value in a range M is determined to reproduce the plot density of a contact map based on a protein 3D structure constructed with the Cα-Cα cutoff distance of 15 Å). In this way, the ADM for a protein can be constructed from the sequence without any information on 3D structure. An example of ADM for hen egg white lysozyme is shown in Figure 2. An area with a high density of plots can be regarded as a region predicted to form a compact unit in the 3D structure. The detailed procedure to detect such a region is described in Supplementary Information 2. An index of a region with high density plots is the η-value. We use the η-value of a region as a measure of strength of compactness. We regard such a region predicted by ADM as a compact region forming in the early stage of folding. In HEWL, there are three regions with high η-values, and thus its ADM predicts the three compact regions in this protein.

Figure 2: The average distance map (ADM) of HEWL. A region enclosed by a red triangle represents the predicted compact region by ADM. A grey bar and an arrow on the diagonal line mean α-helix and β-strand, respectively
We have confirmed that the predicted regions by ADMs for myoglobin and plant leghemoglobin [8], fatty acid binding proteins [8] and ferredoxin-like fold [12] proteins correspond well to the early folding regions detected by NMR studies [23,24], kinetic studies [25] and η-value analyses [26-29].
Comparison of the regions predicted by two ADMs
The similarity of the location of predicted compact regions by ADMs for two sequences is useful information in regard to molecular evolution. The similarity of two ADMs is defined as follows. Suppose that a multiple sequence alignment is obtained.
1. Two sequences are chosen from the aligned sequences.
2. A site with a gap in either one or both sequences is out of consideration. Here, “site” is referred to the common sequential number in the multiple alignments.
3. The number of sites that are commonly included in or excluded from the regions predicted by the ADMs is calculated. The ratio of this number to the number of all the non-gapped sites is defined as the identity of two ADMs. Gapped sites are not taken into consideration and thus the present method is not suitable for alignment including sequences with large gaps. A η-value for a region predicted by ADM does not affect the comparison of two regions.
Contact frequency analysis using a potential derived from the inter-residue average distance statistics
Contact frequency of a residue with other residues in a random state is estimated using a potential derived from the present inter-residue average distance statistics in order to determine the location where initial folding events, such as hydrophobic collapse, happen [8].
In the present analysis, we employed a Cα bead model to represent a protein’s structure. For a simulation of protein conformations the Metropolis Monte Carlo method with the potential energy εi,j derived from average distance ͞r i,j and its standard deviation σi,j was used. The bond and dihedral angles of an initial conformation were randomly chosen.
During a simulation, the bond and dihedral angles between the residue i and i+1 are bent and rotated randomly followed by the Metropolis judgment to decide whether the new conformation can be accepted or not. That is, we started a simulation with totally random distribution with the restriction derived from the average distance statistics. One step includes residues i=1…N-1, that is, all the bond and dihedral angles are altered and judged.
It is assumed that the probability density with the potential energy between two residues, P(εi,j), is equivalent to the probability density derived from the standard Gaussian distribution calculated with its average distance and standard deviation, 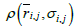 , as follows:
, as follows:
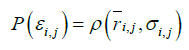 (1)
(1)
This equation can be expressed by equation (2);
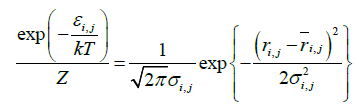 (2)
(2)
Equation (2) leads to equations (3) and (4):
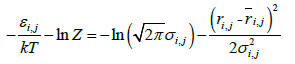 (3)
(3)
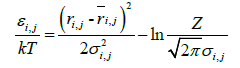 (4)
(4)
Where kT is set so that the acceptance ratio is 0.5. Thus, this potential is expected to obtain ensembles reproducible in regard to inter-residue average distance statistics. It is noted that a significant value is the difference between those of two conformations and Z does not appear in the calculation explicitly. Thus, Z is ignored in calculations. From the results of simulations, the contact frequency, g(i,j), for each pair of residues is calculated with sampled structures generated using the potential energy function. Then we normalize the residue contact frequencies, g(i,j), in the same range M as follows:
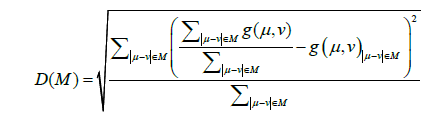 (5)
(5)
 (6)
(6)
Where μ or v is a residue number.
Finally, we obtain the relative contact frequency, Fi, by summing the normalized contact frequencies, Q (i,j), from j=1 to N for each residue i, where N is the total number of residues:
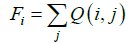 (7)
(7)
We call Fi the F-value. Residues at peaks of the plot of F-values are expected to be located in the center of many inter-residue contacts, such as at a hydrophobic cluster. A region around a peak in an F-value plot is plausible to be significant for folding, especially at the initial stage. We performed 10 simulations with 60000 steps, calculating the average of the F-values for residue i (We calculate the sampled structure from the very begging of the simulation).
We attached a sequence of 10 glycine to both N- and C-termini to avoid too dynamic motions of residues at both ends.
A peak is defined when the difference in the values of a valley and a peak is more than the following cut-off value, Fcut:
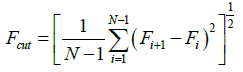 (8)
(8)
Where Fi is the F-value of residue i, and N is the total residue number.
We have confirmed that a hydrophobic residue near the F-value plot for a protein tends to form hydrophobic packing in the native structure of a protein for IgG binding domain [7] and ferredoxin-like fold proteins [12].
Evolutionary analysis
It has been confirmed that evolutionarily conserved residues in a protein are responsible for its function, stability and/or structural formation [30-35]. In this study, we attempt to identify conserved hydrophobic residues. We regard the following residues as hydrophobic residues; Ala, Phe, Ile, Leu, Met, Val, Tyr, and Trp. When 90% of residues at an aligned site are hydrophobic in a multiple alignment, this site is regarded as an “evolutionarily conserved hydrophobic site”.
MAFFT [36] was used to align obtained sequences to investigate evolutionary conservation of predicted regions. We also constructed molecular phylogenetic trees by the Neighbor Joining method incorporated into MEGA ver 7.014 [37] to examine evolutionary relationships among predicted regions in this study. The amino acid substitution matrix used for construction of a phylogenetic tree is the Jones-Taylor-Thornton model which is widely used for this purpose [38]. Complete deletion was used as the gap data treatment option. We checked the bootstrap values obtained from 1000 replications for the topology of a phylogenetic tree. If a value is not extremely low, we regard this topology as valid.
Structural alignment
We made a multiple sequence alignment based on 3D structures using the Combinatorial Extension (CE) program [39] integrated within the STRAP software [40] in order to identify structurally common regions among query structures.
Analyses of queries of lysozyme families based on inter-residue average distance statistics
We describe the results on the ADM and F-value analyses for each lysozyme protein used as a query for BLAST search in this section. The regions predicted by ADM are summarized in Table 2. The details of the positions of peaks of the F-value plot are presented in Table 3.
| Protein Name | Predicted Regions by ADM (η-value) | Dominant Side |
|---|---|---|
| HEWL | Lys1-Ala32 (0.354), Ile55-Cys64 (0.235), Leu75-Cys127 (0.201) |
N |
| BLA1 | Val8-Cys61 (0.192), Ile72-Cys111 (0.225) | C |
| TjL | Ile25-Cys49 (0.210), Ile63-Val122 (0.330) | C |
| GL | Arg1-Ile70 (0.284), Ile119-Ile127 (0.208), Trp134-Tyr180 (0.160) |
N |
| LaL | Phe4-Ile17 (0.008), Lys35-Asn55 (0.205), Ala93-Arg125 (0.219), Tyr139-Phe153 (0.009) |
C |
| 1Bovine α-lactalbumin | ||
Table 2: The summary of location of the predicted regions by ADM in the query sequence in each family. “Dominant side” means whether the predicted region showing the highest η-value locates in the N-or C-terminal side.
| F-Value | |
|---|---|
| HEWL | 11, 16, 26, 29, 31, 36, 54, 59, 63, 68, 72, 77, 88, 92, 107, 110 |
| BLA | 25, 28, 40, 42, 53, 59, 73, 75, 81, 84, 90, 96, 99, 102 |
| TjL | 13, 22, 25, 28, 36, 38, 45, 61, 67, 70, 78, 83, 85, 101, 105, 118 |
| GL | 5, 8, 12, 15, 20, 26, 32, 36, 38, 53, 55, 60, 66, 68, 77, 79, 81, 85, 89, 92, 94, 108, 110, 114, 118, 133, 146, 149, 151, 154, 157, 161, 163, 171, 174 |
| LaL | 11, 14, 21, 36, 41, 42, 46, 54, 70, 73, 94, 106, 108, 113, 119, 135, 140, 148, 152 |
| The Protection Factor for Native State | |
| HEWL [42] | 12, 29, 52, 83, 95 |
| BLA [46] | 12, 27, 42, 62, 75, 93 |
| LaL [48] | 20, 36, 68, 94, 107, 118, 147 |
| The Protection Factor for Molten Globule State | |
| BLA [46] | 9, 26, 41, 53, 61, 75, 92 |
Table 3: The summary of the positions of the peaks of the F-value plots and the major peaks of protection factor plots.
Figure 3 shows the results of ADM and F-value analyses for HEWL with the protection factor values of the H/D exchange experiment with NMR for the native state by Radford et al. [41]. The positions of major peaks of the H/D protection factor in Figure 3 are also presented in Table 3. The ADM analysis predicts the region includes two N-terminal helices (Lys1-Ala32, η=0.354), the region around β3 (Ile55-Cys64, η=0.235), and the region contains C-terminal helices (Leu75-Cys127, η=0.201). The result of the H/D exchange protection [41] for the fluctuation of the native state shows the high protection at two N-terminal helices, α1 and α2 (the name of a secondary structure is shown in Figure 1), reflecting the higher η-value of the first predicted region, that is, the predicted region with the higher η-value by ADM corresponds to the part structured well with relatively low fluctuation in the native state of HEWL. Radford et al. [42] also demonstrated that α1 and α2 show faster protection in the early stage of folding consistent with the result of ADM. The peaks of the protection factor histograms for each state correspond well to the peaks in the F-value plot as shown in Figure 3. Table 3 and Figure 3 show that a peak in the F-value plot is located near the maximum peak of a cluster of high values of an H/D protection factor histogram within ±5 residues. It is interesting that the protection factor obtained from the refolding experiment for equine lysozyme shows the higher peaks at α1 and α2 [43]. Similar results were obtained for human lysozyme in a refolding experiment [43]. Thus, we consider that the ADM and F-value analyses predict the folding properties of HEWL well.

Figure 3: The F-value plot for HEWL. The grey histogram expresses the protection factor of the native state [41]. A grey bar and an arrow in the bottom of a plot mean α-helix and β-strand, respectively. A black bar in the bottom of a figure represents a predicted region by ADM. “Primary” means the region with higher η-value. A red dot indicates an evolutionarily conserved hydrophobic residue. A thin arrow denotes a major peak in the F-value plot. A broken arrow denotes a small peak in the F-value plot.
Figure 4A and 4B represents the results of the present analyses with the protection factor values of H/D exchange experiment by NMR for both the native and molten globule states [44] of bovine α-lactalbumin (BLA), a homologue of C-type lysozyme. This experimental data of the H/D exchange protection reveals that the region around α3 structures is highly protected in the molten globule state [45]. Our ADM prediction shows that the predicted unit is Glu1-Cys120, which is 98.3% of the whole sequence. Therefore, we made another prediction for the truncated sequence, Val8-Cys111 (ADM predicts this region as a possible compact region as a whole. ADM of BLA is shown in Supplementary Information 3.) As a result, the N-terminal helical region with the sheet region, Val8-Cys61 (η=0.192), and the C-terminal helical region, Ile72-111 (η=0.225), are predicted. High values of the C-terminal residues (around α3) are observed in the protection factor histogram and this fact corresponds well to the higher η-value of the C-terminal region predicted by ADM. The good correspondence can be made between the location of the peaks of the F-value plot and that of the peaks of the protection factor histogram as observed in Figure 4 and Table 3. For BLA, a peak in the F-value plot is also near the maximum peak of a cluster of high values of the H/D protection factor histogram within ±3 residues. Again our results reflect the experimental data.

Figure 4: The F-value plot for BLA. The grey histogram expresses the protection factor of (A) native state, (B) molten globule state [45]. A grey bar and an arrow in the bottom of a plot denote α-helix and β-strand, respectively. A black bar in the bottom of a figure represents a predicted region by ADM. “Primary” means the region with higher η-value. A red dot indicates an evolutionarily conserved hydrophobic residue. A thin arrow denotes a major peak in the F-value plot.
From the comparisons of the results of the NMR H/D protection factor experiments with those of the present analyses for HEWL and BLA, we consider that a predicted region with higher η-value is assumed to be strong folding initiation segment (folding unit), and a residue around a peak in the F-value plot would also be significant for the folding [We examined the present analyses further for canine milk lysozymes and goat α-lactalbumin, equine lysozyme, human lysozyme, human α-lactalbumins (C-type lysozyme family proteins) [5,42,45,46]. The details are described in the Supplementary Information 4 and 5].
Figure 5 presents the results of ADM and F-value analyses for TjL as a query of I-type lysozyme family. The predicted unit by ADM is Cys10- Val122, 91.0% of the whole sequence. That is, this protein is similar to BLA. Therefore, we made another prediction for the truncated sequence, Ile25-Val122 (ADM predicts this region as a possible compact region as a whole. ADM of TjL is shown in Supplementary Information 3.) The result of the present ADM analysis detects the N-terminal region covering from α1 to α2 (Ile25-Cys49, η=0.210) and the C-terminal region containing α3 to α5 (Ile63-Val122, η=0.330), and thus the C-terminal region shows higher η-value. The peaks in the F-value plot locate in α1, β2-β3, 2, α3, and α5, respectively as shown Figure 5. This result suggests the C-terminal region as the strong folding initiation site. To our best knowledge, there is no experimental data on the folding of this protein. Therefore, we do not make further comparison of our present results to any experimental data.

Figure 5: The F-value plot for TjL. A grey bar and an arrow in the bottom of a plot denote α-helix and β-strand, respectively. A black bar in the bottom of a figure represents a predicted region by ADM. “Primary” means the region with higher η-value. A red dot indicates an evolutionarily conserved hydrophobic residue. A thin arrow denotes a major peak in the F-value plot.
In Figure 6, the results of ADM and F-value analyses for GL as a query of G-type lysozyme family are represented. There is also no experimental data on the folding of this protein. The ADM analysis predicts the N-terminal region covering from α1-α4 (Arg1-Ile70, η=0.284) and the region around α5 (Ile119-Ile127, η=0.208), and the C-terminal region covering from α6-α7 (Trp134-Tyr180, η=0.160), and thus the N-terminal region shows higher η-value. The peaks in the F-value plot locate in α1, α2, α3, α4, α5, α6, α7, and near the β-sheet, respectively. This result suggests the N-terminal region as the strong folding initiation site.

Figure 6: The F-value plot for GL. A gray bar and an arrow in the bottom of a plot denote α-helix and β-strand, respectively. A black bar in the bottom of a figure represents a predicted region by ADM. “Primary” means the region with higher η-value. A red dot indicates an evolutionarily conserved hydrophobic residue. A thin arrow denotes a major peak in the F-value plot.
Figure 7 shows the present analyses with the protection factor values of the H/D exchange experiment by NMR for the native state (pH=5.6, 20°) of lysozyme from λ-phage [47]. It was observed from this experiment that β3 and β4 are highly dynamical and totally unprotected regions.

Figure 7: The F-value plot for LaL. The gray histogram expresses the protection factor of the native state [47]. A grey bar and an arrow in the bottom of a plot denote α-helix and β-strand, respectively. A black bar represents a predicted region by ADM. “Primary” means the region with higher η-value. A red dot indicates an evolutionarily conserved hydrophobic residue. A thin arrow denotes a major peak in the F-value plot.
The ADM analysis predicts the N-terminal region covering α1 (Phe4-Ile17, η=0.008), the region covering from β1 to β3 (Lys35-Asn55, η=0.205), the region covering from α3 to α5 (Ala93-Arg125, η=0.220), and the region around α6 (Tyr139-Phe153, η=0.009), that is, the region from α3 to α5 shows higher η-value. This result suggests the C-terminal α3 to α5 region as the strong folding initiation site. The peaks in the F-value plot locate in α1, β1, β2, β5, β6, α2, α3, α4, and α5, respectively. The highest peak in the F-value plot is located in the region covering β5 to β6, and α2, but there is no region predicted by ADM. We will discuss the role of this region including β5 to β6, and α2 for the folding later. However, the location of the peaks in the F-value plot corresponds well to the peaks of the protection factor histogram within ±1 residue as observed in Table 3 and Figure 7. We also think that a valley around β3 corresponds to the dynamical region observed at β3 and β4 in the experiment by Paolo et al. [47].
Evolutionary conservation of predicted folding units for proteins in each family
The evolutionary conservation of predicted folding units for proteins in each family is examined by multiple sequence alignments with ADM results for homologues in C-type, I-type, G-type, and L-type families obtained by BLAST search. Figures 8-11 show the multiple alignments with predicted units by ADMs and the histogram indicating the ratio of the number of residues at an aligned site which is included in predicted regions to the total number of aligned sequences is shown in the bottom of each figure. The details of this procedure are described in the Supplementary Information 6. The higher this ratio is, the more a folding unit tends to be conserved during evolution. Thus, such a region may constitute a universal folding unit in a family.

Figure 8: The multiple alignment with ADM predictions for C-type lysozyme family. The histogram showing in the bottom indicates the conservation ratio of a predicted region in each site of the multiple sequence alignment. A grey bar and an arrow above of the figure mean α-helix and β-strand, respectively. A red dot and yellow characters in a site of the multiple sequence alignment express conserved hydrophobic residues of this site. A red square bracket at the lower part of the phylogenetic tree indicates the α-lactalbumin cluster. Each α-lactalbumin is denoted by a blue dot.

Figure 9: The multiple alignment with ADM predictions for I-type lysozyme family. The histogram showing in the bottom indicates the conservation ratio of a predicted region in each site of the multiple sequence alignment. A grey bar and an arrow above of the figure mean α-helix and β-strand, respectively. A red dot and yellow characters in a site of the multiple sequence alignment express conserved hydrophobic residues of this site.

Figure 10: The multiple alignment with ADM predictions for G-type lysozyme family. The histogram showing in the bottom indicates the conservation ratio of a predicted region in each site of the multiple sequence alignment. A grey bar and an arrow above of the figure mean α-helix and β-strand, respectively. A red dot and yellow characters in a site of the multiple sequence alignment express conserved hydrophobic residues of this site.

Figure 11: The multiple alignment with ADM predictions for L-type lysozyme family. The histogram showing in the bottom indicates the conservation ratio of a predicted region in each site of the multiple sequence alignment. A gray bar and an arrow above of the figure mean α-helix and β-strand, respectively. A red dot and yellow characters in a site of the multiple sequence alignment express conserved hydrophobic residues of this site.
Table 4 presents a summary of the pairwise sequence identities and ADM identities. The average values of pairwise sequence identities are about 40-50% and those of ADM identities are about 65-75%. Direct comparisons between sequence and ADM identities should be done carefully but these results seem to indicate that ADMs are more conserved than sequences.
| C-type | I-type | G-type | L-type | |
|---|---|---|---|---|
| Average sequence identity (%) | 44.5 | 42.7 | 51.0 | 50.7 |
| Maximum sequence identity (%) | 88.5 | 86.6 | 87.8 | 89.8 |
| Minimum sequence identity (%) | 25.0 | 17.4 | 24.7 | 25.4 |
| Average ADM identity (%) | 68.5 | 65.1 | 73.4 | 69.1 |
| Maximum ADM identity (%) | 100.0 | 100.0 | 100.0 | 100.0 |
| Minimum ADM identity (%) | 15.3 | 7.14 | 27.6 | 29.0 |
Table 4: The summary of the sequence and ADM identities for each family protein within each family.
The results of the multiple alignments with ADM predictions for C-type lysozyme family are indicated in Figure 8. The brighter red color is in the predicted regions, the higher η-value is. We recognize that the regions with higher η-values tend to distribute in the N-terminal parts from Figure 8. This figure includes 72 sequences of C-type lysozyme family collected by the BLAST search. The histogram for the homologues suggests that N-terminal helical, sheet, and C-terminal helical regions tend to be conserved. Homologues of α-lactalbumin form a cluster (each member in this cluster is indicated by a blue dot) in Figure 8 separated from lysozyme homologues, and it is recognized that two folding units are mainly predicted by ADMs. The N-terminal folding unit includes the N-terminal helices and β-sheet and the C-terminal folding unit contains the C-terminal helices as in the case of BLA. The ratios of conserved predicted folding units exceed 70% for the regions Cys6-Ala32, Ile55-Cys64, and Leu75-Trp111 in the sequence of HEWL. The conservations of hydrophobic residues are observed at 25 sites in the multiple alignments as indicated in Figure 8. In this figure, these sites are indicated by red points in the upper part of the figure. The conserved residues are coloured by yellow in the predicted regions and otherwise blue in this figure. If we regard a region with more than 70% ratio of conservation of predicted regions tentatively as evolutionarily conserved predicted unit, then 21 out of the 25 conserved hydrophobic residues are included in these units as presented in Table 5 and Figure 8. Furthermore, 19 of these 21 residues are near the peaks of the F-value plot for HEWL.
| Whole Sequence | Predicted Region of Query | Highly (≥70%) Conserved Region | |
|---|---|---|---|
| C-type | 25 (20) | 23 (19) | 21 (19) |
| I-type | 14 (13) | 11 (10) | 11 (10) |
| G-type | 41 (37) | 33 (29) | 27 (23) |
| L-type | 35 (25) | 24 (23) | 20 (19) |
Table 5: The number of evolutionarily conserved residues located on whole sequence, predicted region of query sequence, and highly conserved region in each family. The number in a parenthesis is the number of conserved residues near the peaks of the F-value plots
The lysozyme and α-lactalbumin from echidna show strange properties of the regions predicted by ADMs. Echidna milk lysozyme is quite close to echidna α-lactalbumin within the cluster of α-lactalbumins on the phylogenetic tree. This is only one lysozyme in the cluster of α-lactalbumin. The ADM predicted regions for Echidna milk lysozyme are Lys1-Cys64 (η=0.386), and Ala75-Trp111 (η=0.181) which are close to other α-lactalbumins rather than lysozymes. On the other hand, those for Echidna α-lactalbumin are Lys1-Phe33 (η=0.386), Leu54-Cys63 (η=0.197), and Val75-Ala119 (η=0.167), and those are close to other lysozymes rather than α-lactalbumin. These phenomena may suggest the inter-compatibility of the functions and folding mechanisms during the evolution between α-lactalbumin and lysozyme.
Figure 9 presents the multiple sequence alignment with ADM predictions of the TjL and its homologues. The BLAST search for I-type lysozyme query sequence hit 52 sequences, and these sequences are included in this figure. We see from the brightness of red bars that the η-values of predicted regions are not so much different between the N-and C-terminal regions. The N-terminal α1-α2 with β-strands and the C-terminal helical regions tend to be included in the predicted folding units. The ratios of conserved predicted folding units of the histogram exceed 70% for the regions α1, β1-β3, α2, α4 and α5. The corresponding regions in TjL are Cys13-Cys49, Cys59-Ala78, and Arg82-Arg105. Eleven out of the 14 conserved hydrophobic residues are included in the evolutionarily conserved units as shown in Table 5 and Figure 9. Furthermore, 10 of these 11 residues are near the peaks in the F-value plot for TjL.
Multiple alignments with ADM predictions for G-type lysozymes are shown in Figure 10 including 53 sequences. The η-values for folding units in each protein tend to be large in C-terminal units. As the results of ADM analyses, predicted folding units tend to cover α2-α3, α3-α4 in N-terminal region and α5-α6 and α7 in C-terminal region in G-type lysozyme. α2, α3, α4, α5, α6, and α7 are located in the regions with more than 70% ratio of conserved predicted folding units of the histogram. These regions correspond to the regions Cys18-Ala38, Ile52-Ile70, Ile119-Ile127, and Ile144-Tyr180 in the GL. Furthermore, the regions corresponding to β1-β2 show moderate folding unit conservation. Twenty seven out of the 41 conserved hydrophobic residues are included in the evolutionarily conserved units as shown in Table 5 and Figure 10. Furthermore, 23 of these 27 residues are near the peaks in the F-value plot for GL.
In the same way, multiple alignments with ADM predictions for 100 L-type lysozymes are shown in Figure 11. The C-terminal predicted region in each protein tends to show higher η-value. Unexpectedly, the present BLAST search hit only lysozymes from bacteria in spite of the fact that lysozyme from bacteriophage was used as a query. The results of ADM analyses indicate that predicted folding units tend to cover from β1-β2, 3-α5, and α6. β1, β2, α3, α4, α5, and α6 are included in the regions more than 70% ratio of conserved predicted folding units of the histogram. These regions correspond to the regions Tyr33- Phe42, Val94-Ala125, and Gly129-Phe146 in the LaL. Furthermore, the regions corresponding to α1, β3-β6 and α2 imply moderate folding unit conservation. Twenty out of the 35 conserved hydrophobic residues are included in the evolutionarily conserved units as shown in Table 5. Furthermore, 19 of these 20 residues are near the peaks in the F-value plot for LaL.
As demonstrated above, as shown in Table 6, the locations of evolutionarily conserved units among homologues coincide with the regions predicted by the ADM for the query in each family. β1-β2 in G-type lysozymes and β3-β4, β5-β6, 2 in L-type lysozymes show moderate conservation in the histograms. These are not predicted in the ADMs for the queries.
| Highly (≥70%) Conserved Regions | Modestly Conserved Regions | |
|---|---|---|
| C-type | α1-α 2, b2-b3, α3-α5 | α6 |
| I-type | α1-α2 (with b), α3-α5 | - |
| G-type | α1-α4, α5, α6-α7 | b1-b2 |
| L-type | b1-b2, α3-α5, α6 | α1, b3-b4, b5-b6, α2 |
Table 6: The correspondence of the secondary structures to the location of highly conserved regions and modestly conserved regions.
Comparisons of predicted folding cores by ADM and F-value plot to 3D structure
As mentioned previously, it is observed that the regions predicted by ADM in each query protein are conserved during evolution. This finding suggests that the folding mechanism of each query protein might be conserved during evolution. In particular, conserved hydrophobic residues in a predicted region might be involved in the formation of the folding core significantly. To confirm this, we analyse the distribution of the predicted folding units and the interactions formed by conserved hydrophobic residues in the 3D structure of a query protein.
Table 7 shows the number of hydrophobic contacts between conserved hydrophobic residues in each lysozyme. This table suggests that the major part of the contacts is constituted within the predicted units for each lysozyme. (All contacts formed by conserved hydrophobic residues are presented in Supplementary Information 7.) Figure 12A-12D show the distribution of predicted units and conserved hydrophobic residues located near the peak in the F-value plot within ±5 residues in the 3D structure of HEWL, TjL, GL, and LaL. (The average (ā) and the standard deviation (σ) values of the deviation between the peaks in the H/D protection factor histograms and the F-value plots are 1.70 and 1.54 calculated from Table 3 and Table S3. As a result, ā+ 2σ=4.78. A peak in an F-value plot exists near a peak in an H/D protection factor histogram within around ±5 residues with 95% of statistical significance.) In each figure, predicted units are distinguished by different colors, and a residue near a peak in the F-value plot is expressed by a CPK space filling molecular model of the protein. Figure 13A-13D show the predicted units and conserved hydrophobic residues far from the F-value peaks (>5 and <-5) for each lysozyme. Such residues are coloured magenta. For each lysozyme, it is recognized that these conserved hydrophobic residues locate on the surface of the protein (far from the core) and are not involved in the hydrophobic packing in Figure 13. Thus, it is confirmed for each lysozyme that the conserved hydrophobic residues near peaks of the F-value plot have a significant role forming the hydrophobic core.
| Unit1 | Unit2 | Unit3 | Unit4 | ||
|---|---|---|---|---|---|
| HEWL | Unit1 (α1-α2) | 28 | 6 | 13 | - |
| Unit2 (b2-b3) | 6 | 6 | 16 | - | |
| Unit3 (α3-α6) | 13 | 16 | 28 | - | |
| TjL | Unit1 (α1, b1-b3, α2) | 8 | 5 | - | - |
| Unit2 (α3-α5) | 5 | 16 | - | - | |
| GL | Unit1 (α1-α4) | 48 | 3 | 19 | - |
| Unit2 (α5) | 3 | 6 | 8 | - | |
| Unit3 (α6-α7) | 19 | 8 | 48 | - | |
| LaL | Unit1 (α1) | 26 | 0 | 12 | 5 |
| Unit2 (b1-b2) | 0 | 6 | 0 | 0 | |
| Unit3 (α3-α5) | 12 | 0 | 42 | 3 | |
| Unit4 (α6) | 5 | 0 | 3 | 2 |
Table 7: Number of contacts formed by conserved hydrophobic residues within each unit and inter-units.

Figure 12: The location of predicted folding unit in the 3D structures of (A) HEWL, (B) TjL, (C) GL, and (D) LaL. A folding unit in each figure is distinguished by color. The conserved hydrophobic residues near the peaks in the F-value plots are shown by the CPK model. Tyr67 in LaL is emphasized by black color.

Figure 13: The conserved hydrophobic residues (magenta) located at the region far from the peaks in the F-value plots for (A) HEWL, (B) TjL, (C) GL, and (D) LaL. Packing residues near the peaks in the F-value plots are indicated with CPK model with semi-transparent colours.
For LaL, it is worthwhile to mention that Tyr67 is not in a predicted unit but this is near one of the peaks in the F-value plot and interacts with conserved hydrophobic residues existing in units 1, 2 and 3. That is, Tyr67 seems to link units 1, 2 and 3.
Multiple alignments of 3D structures of lysozymes
Figure 14 represents the multiple alignments of 3D structures of the query lysozymes by the Combinatorial Extension (CE) method [40] with the regions predicted by the ADMs. In this figure, aligned local sequences denote the structurally similar portions in these proteins.

Figure 14: The multiple alignments of query proteins based on 3D structures. Aligned regions for these proteins denote that the 3D structures of the parts that are similar. A white bar indicates each secondary structure (labelled by black characters). A secondary structure with red characters denotes a portion that is observed in all four queries. These secondary structures include the conserved hydrophobic residues near the peaks in each F-value plot. A red bar on the sequence for each query denotes a predicted region by ADM.
The correspondence of the secondary structures among four lysozymes are as follows: “α2, α1, α4, and α1”, “β2-β3, β2-β3, 1-β2, and β5-β6”, “α4, α3, α5, and α3”, “N-terminus of α5, α4, α6, α5”, and “C-terminus of α5, C-terminus of α5, α7, α6” in order of HEWL, TjL, GL, and LaL as indicated in Figure 14. Furthermore, we can make correspondence between these aligned local regions in Figure 14 and the segments identified visually in Figure 1.
Thus, these five regions in each lysozyme can be regarded as structurally common regions among all four lysozymes. Figure 15 shows the conserved hydrophobic residues near the peaks in the F-value plots in the structurally common regions in each lysozyme, and a residue in a structurally common region makes a hydrophobic contact with another region in a lysozyme (Details of the hydrophobic contact residues are presented in Table 8).

Figure 15: The location of the contacts between conserved hydrophobic residues near the peaks in the F-value plots in the structurally common regions on the 3D structures of (A) HEWL, (B) TjL, (C) GL, and (D) LaL.
| HEWL (PDB ID: 2VB1) | TjL (PDB ID: 2DQA) | |
|---|---|---|
| C1⇔C2 | Trp28-Leu56 | Leu11-Phe39, Met14-Phe39 |
| C1⇔C3 | Trp28-Ala95 | Leu11-Val70, Met14-Val70 |
| C1⇔C4 | Trp28-Met105, Trp28-Trp108 | Met14-Phe90 |
| C2⇔C3 | Ile55-Ile88, Ile55-Val92, Leu56-Val92, Ile58-Ala95 | Phe39-Val70, Ile41-Val70 |
| C2⇔C4 | Leu56-Met105, Leu56-Trp108, Ile58-Trp108 | None |
| C3⇔C4 | Ala95-Trp108 | Met74-Phe90 |
| GL (PDB ID: 153L) | LaL (PDB ID: 1AM7) | |
| C1⇔C2 | Ile66-Met94, Ile69-Leu93, Ile69-Met94 | Leu12-Tyr67, Leu15-Tyr67 |
| C1⇔C3 | Val65-Leu120, Ile69-Leu120 | Phe11-Ile99, Leu12-Ala95, Leu12-Leu96, Leu12-Ile99, Leu15-Ile99 |
| C1⇔C4 | Val65-Ile144, Ile69-Ile144, Ile69-Tyr147 | Phe11-Ile113, Phe11-Ala116, Phe11-Ile117, Met14-Ile113, Met14-Ile117 |
| C2⇔C3 | Leu93-Ile113, Met94-Ile113 | Tyr67-Ala95 |
| C2⇔C4 | Met94-Tyr147, Val96-Tyr147 | None |
| C3⇔C4 | Ile119-Tyr147, Leu120-Ile144, Leu120-Tyr147, Ile124-Ile144 | None |
Table 8: The contacts between conserved hydrophobic residues near the peaks of the F-value plots in structurally common regions (C1, C2, C3 and C4).
It is notable in this figure that conserved hydrophobic residues included in the structurally common regions 1, 2, and 3 (we call these regions C1, C2, and C3) occur close to each other in all query proteins. Contacts of conserved hydrophobic residues in structurally common regions 1 and 4 (C1 and C4) are also observed within four query lysozymes. In HEWL, TjL, and GL, there are some contacts of conserved hydrophobic residues in structurally common regions 3 and 4 (C3 and C4). These conserved residues are summarized in Table 8, and the contacts mentioned above can be regarded as common contacts among the structurally common regions in four lysozymes.
The fifth structurally common region does not contain a conserved hydrophobic residue near a peak in the F-value plot for each lysozyme, and thus it is speculated that this part is not involved in the formation of the common topology in each lysozyme.
It is confirmed in Figures 8-10 that structurally common regions correspond well to one of the well-conserved predicted regions (region with more than 70% ratio of conservation of predicted regions) in C-, I-, and G-type lysozymes, although the conservation of the sheet region in G-type lysozymes is somewhat modest as shown in Figure 10. In L-type lysozymes, the correspondence between structurally common regions and conserved predicted regions is not so remarkable. Conserved predicted regions include β1-β2, α3-α6 and among them, structurally common regions are only α3 and α5. This suggests that the folding mechanism of an L-type lysozyme is rather different from that of other lysozymes. We will come to this problem in discussion.
It should be noticed that two conserved hydrophobic residues in structurally common regions forming a contact are always on major peaks in an F-value plot as shown in Figures 3-7 and 15.
As we mentioned previously, analyses based on average distance statistics reproduce well the folding properties of C-type lysozymes and α-LaL. The results obtained in this study and our preceding studies amply demonstrate that among predicted compact regions by ADM, a region with a high η-value can be regarded as a region with high contact density and stability in the early stage of folding. In particular, hydrophobic residues conserved during evolution near the peaks in the F-value plot of a protein can be regarded as residues highly involved in forming contacts within this region and inter-regions.
The conservation of the predicted folding units obtained by the present evolutionary analyses (Figures 8-11) suggests the conservation of folding mechanisms in each family. It is interesting to further examine of the correlation between ADM and sequence identities presented in Table 4. Figure 16A-16D shows the plots of these values for pairs of sequences in families. These figures show that there is no remarkable correlation between them for each lysozyme family. However, there are sequence pairs which show high ADM identity in spite of low sequence identity (indicated by the red rectangles in Figure 16A-16D and this fact suggests that two distantly relate proteins in evolution show similar folding mechanisms. In other words, it seems rather difficult to discuss similarity of folding mechanisms of two proteins just based on sequence identity. These results suggest that the folding mechanisms between lysozymes from different families with low sequence identity can nevertheless be similar to each other.

Figure 16: The scattered plots between sequence identity and ADM idensity in (A) C-type, (B) I-type, (C) G-type, (D) L-type lysozyme family. A red rectangle denotes the cluster of pairs of sequences of proteins in each family denoting high ADM identities (80-100%) in spite of relatively low to modest sequence identity (20-40%).
The regions predicted by ADMs for C-type (especially α-lactalbumin cluster) and I-type lysozymes resemble each other as seen in Figures 8 and 9. Based on this result with the relatively high similarity of the 3D structures between HEWL and TjL, we can infer the high similarity of general folding mechanisms of C-type and I-type lysozymes. Similar correspondence of the results of C-type and G-type lysozymes can be observed. In particular, the similarity of the regions predicted by ADMs for C-type and G-type lysozymes is observed in Figures 8 and 10 except the moderate conservation around β1 and β2 in G-type lysozymes. These similarities of the predicted folding units among C-type, I-type, and G-type lysozymes suggest the similarity of the gross features of general folding mechanisms of lysozymes of the animal kingdom.
Furthermore, multiple alignments based on the 3D structures (Figure 14) reveal the structurally common regions among the four query lysozymes. These regions are consistent with the common regions intuitively obtained (Figure 1). We confirmed that four of the structurally common regions contain peaks in the F-value plots (Figures 3-7). These structurally common regions tend to be in the evolutionarily conserved folding unit.
We point out the common hydrophobic interactions connecting the evolutionarily conserved folding units for four lysozymes as shown in Table 8, and Figures 12A-12D and 15A-15D.
These facts suggest that these common hydrophobic interactions are conserved during evolution. Thus, we think that the partially common topology of each lysozyme is formed by these interactions. It is plausible that these common hydrophobic interactions are indispensable for forming the partially common topology in lysozymes. Furthermore, we consider these contacts are evolutionarily conserved.
It is interesting how the common structure pointed out in this study relates to the common function. The functional residues of HEWL are identified as Glu35, Asp52 [15], and those of TjL are Glu18, Asp30 [1,15]. In GL and LaL, Glu73 and Glu19 are recognized as the functional residues [2,3,15]. However, a corresponding Asp is not identified in these proteins. Wohlkonig et al. [4] pointed out that the secondary structures responsible for the function are a helix including functional Glu and a β-hairpin near the substrate binding site are evolutionarily common among this superfamily, and this β-hairpin contains potentially important residues for the function. The interesting point is that among the structurally common regions defined in this study, C1 and C2 correspond to this helix and the λ-hairpin. We suggested previously that the contacts between the conserved hydrophobic residues near the peaks in the F-value plots are significant to form the common topology and these contacts can be regarded as significant for the common function.
Lysozyme families contain high variety in their 3D structures. A V-type lysozyme is rather different from other lysozymes. It would be very interesting to study whether the folding mechanism of a V-type lysozyme is similar to that of the lysozymes from other family. We are currently working on this problem.
This work was supported by JSPS KAKENHI Grant Number 26330335.