Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2012) Volume 5, Issue 9
In this paper, proteomics have been studied in the light of Integral Value Transformations (IVTs) which was introduced in. For case study, a Human olfactory receptor OR1D2 protein sequence has been considered as the initial sequence and then different IVTs have been used to evolve OR1D2 into some other proteomic sequences. As ensued, it has been found that some of the generated sequences have been mapped to another olfactory receptor in Human or in some other species. Also it has been corroborated through fractal dimension that some of the fundamental protein properties have been nearly intact. Thus, we propose a methodology via which proteins having the same properties can be connected or grouped. This study will help to comprehend proteomic evolutionary network through IVTs.
Keywords: Olfactory receptors (ORs); Box-counting dimension; Proteomics
The study of proteins such as structures, functions and evolutions is universally known to as Proteomics, was first coined in 1997 to make an analogy with Genomics, the study of the genes [1]. After genomics, proteomics is considered to be the next step in the study of biological systems. While we humans probably have only some 21 thousand genes, we possess at least 10 times that number of different proteins. The study of proteomics is important because proteins are responsible for both the structure and the functions of all living things. Genes are simply the instructions for making proteins. Therefore, a proper quantitative understanding of proteins characteristics and their inter-network are required. In this paper, an olfactory receptor OR1D2 has been considered for our analysis. Interestingly, on applying IVT systematically, we have been able to show that each DNA sequence at various discrete time instances in IVT evolutions can be directly mapped to another specific proteomic sequences existing in different species. A number of the fundamental properties namely percentage of accessible residues, alpha helix (Chou & Fasman), amino acid composition (%), beta sheet (Chou & Fasman), beta turn (Chou & Fasman), coil (Deleage & Roux), hydrophobicity (Aboderin) and total beta strand have been considered for the protein properties of the IVT generated sequences. All protein plots for all the IVT generated sequences including OR1D2 (the primitive sequence) have been generated using MATLAB (bioinformatics toolbox). Then box-counting dimension for each of the protein plots have been calculated through BENOITTM. This study will help us to ascertain potential new drugs for the treatment of various diseases.
In this section, we describe very briefly about IVTs, fractal and proteins.
Notion of integral value transformation (IVT)
Let us define the Integral Value Transformations (IVTs) in N0K as the following [2-5]:
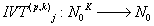
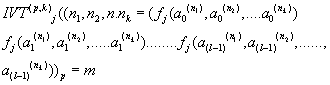
where
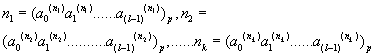
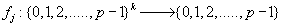
m is the decimal conversion from the p adic number.
Obviously for 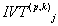 system there are
system there are 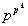 number of possible rules and out of them the function number that we select is indicated by j.
number of possible rules and out of them the function number that we select is indicated by j.
Let us fix the domain of IVTs as N0 (k=1) and thus the above definition boils down to the following:
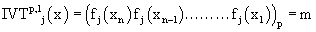
where m is the decimal conversion from the p adic number, and
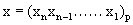
Now, let us denote the set of 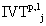 as
as
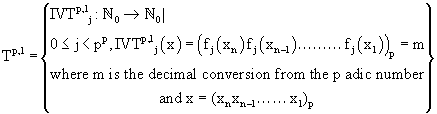
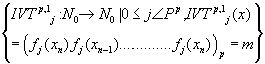
Where m is the decimal conversion from the P adic number and
Let us define the IVT in N0 in 4-adic number systems. There are 256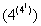 one variable four state CA rules. Corresponding to each of those CA rules there are 256 IVTs in 4 adic system in one dimension.
one variable four state CA rules. Corresponding to each of those CA rules there are 256 IVTs in 4 adic system in one dimension.
IVT4,1 # is mapping a non-negative integer to a non-negative integer.
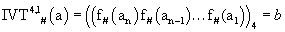
Where ‘a’ is a non-negative integer and 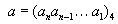 and ‘b’ is the decimal value corresponding to the 4-adic number.
and ‘b’ is the decimal value corresponding to the 4-adic number.
For an example, let us consider a = 225 = (3201)4 and 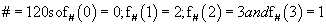
Therefore, 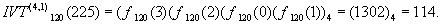
Consequently, 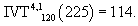
Let us denote T4,1# as set of all IVTp,k # transformations. It is worth noting that there are 4! = 24 number of bijective functions in T4,1# . So out of the 256 transformations in T4,1 # four are linear and rest is nonlinear [6].
Fractal and fractal dimension
Our artificial world can be described easily through Euclidean geometric shapes but there are many things in nature such as shape of cloud, geometry of lightening etc. could not be described through Euclidean geometry. Many mathematicians descended the challenge for a fair enough description of natural objects but after a long period in 1975, B. Mandelbrot took up the challenge and gave the birth of a new geometry to describe nature which is known to us as ‘Fractal Geometry’ (in short ‘Fractal’). The precise definition of "Fractal" according to Benoit Mandelbrot is a set for which the Hausdroff Besicovitch dimension strictly exceeds the topological dimension. To gain a quantitative insight of Fractal, some fractal parameters namely Fractal dimension, Hurst exponent, succolarity, lacunarity etc. are also introduced in the literature. A brief discussion follows about one of the well-known methods of calculating fractal dimension namely ‘Box- Counting method’.
Box-Counting Method: This method computes the number of cells required to entirely cover an object, with grids of cells of varying size. Practically, this is performed by superimposing regular grids over an object and by counting the number of occupied cells. The logarithm of N(r), the number of occupied cells, versus the logarithm of 1/r, where r is the size of one cell, gives a line whose gradient corresponds to the box dimension [7].
Problem in protein structures
Proteins are an important class of biological macromolecules present in all organisms. After the structure of DNA was discovered by James Watson and Francis Crick, who used the experimental evidence of Maurice Wilkins and Rosalind Franklin (among others), serious efforts to understand the nature of the encoding of proteins began. George postulated that a three-letter code must be employed to encode the 20 standard amino acids used by living cells to encode proteins, because 3 is the smallest integer n such that 4n is at least 20 [8]. The three-dimensional structures of proteins were first determined by X-ray diffraction analysis; Perutz and Kendrew shared the 1962 Nobel Prize in Chemistry for these discoveries. At present, more than ten thousand protein structures were found with their atomic details. The structure of the protein is ultimately defined by its primary structure, or amino acid sequence. There are no theories or computational techniques at the moment which will allow us to predict the new protein folding by its sequence. Even, how protein sequences and their tertiary structures are evolved during evolution remains unclear. Therefore proper understanding is required at the primary structure level i.e. in the amino acids sequence level of proteins.
Method of sequence generation through IVTs
The domain of action of IVTs is a set of non-negative positive integers. So it is required to have a numeric sequence corresponding to each of the proteomic sequence. A simple mapping f is defined below:
Let P = {A,C,D,E,F,G,H, I,K,L,Q,N,P,Q,R,S,T,V,W,Y} be the set of amino acid codes and
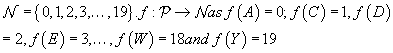
Therefore, a protein sequence is now simply a string of twenty variables namely 0, 1, 2…19 as per coding scheme f.
Starting from a protein sequence to generate another proteomic like sequences, it is required to have all the IVTs in a particular T(p,1)# ,which maps N to itself (bijective rules).
The list of some such IVTs in T(p,1)# is given below in Table 1.
P-adic |
 |
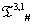 |
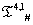 |
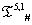 |
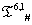 |
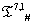 |
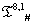 |
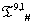 |
|---|---|---|---|---|---|---|---|---|
| # | 1 2 | 5 11 21 | 99 114 147 177 180 210 225 228 | 194 214 294 334 414 434 694 714 894 | 28565 28595 28745 28805 28955 28985 29860 29890 30040 30100 | 297051 297093 297393 297435 299109 299151 299793 299835 | 5135375 5135431 5135886 5135942 5138959 5139015 5139981 | 102907844 102907916 102908572 102908644 102913676 102913748 102915132 |
Table 1: IVTs in .
Now we apply Integral Value Transformations ,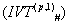 systematically [3-10]:-
systematically [3-10]:-
Firstly, divide the whole one dimensional initial sheet of proteomic sequence (numeric sequence) of length n into multiple blocks. We designate the initial sequence as S(t0 ) .
Secondly, we apply bijective domain preservative transformations (need not to be all distinct) taken from , for different p starting from 2 to 19) over each of the r different blocks in t0. We call such application of different rules to different blocks as Hybrid Application of IVTs. In other words, we are getting S(t1) from S(t0)through hybrid application of IVTs. Next, we follow this step successively as long as we wish to iterate. The results, on applying the proposed systematic technique of application of IVTs on OR1D2 are enumerated in the following subsections.
Here we discuss the results on applying different IVTs in two following cases.
On applying ,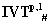 : The proteomic sequence of OR1D2 is of length 312 (sequence shown below in Text-1). Choose r=50, so there are 7 blocks are there. The following two IVTs are used to generate S(t1) as shown below in Table 2.
: The proteomic sequence of OR1D2 is of length 312 (sequence shown below in Text-1). Choose r=50, so there are 7 blocks are there. The following two IVTs are used to generate S(t1) as shown below in Table 2.
| BLOCK | Sequence-1 in 2 adic IVT | Sequence-1 in 3 adic IVT | Sequence-1 in 4 adic IVT |
|---|---|---|---|
| Block-1 | IVT2,11 | IVT3,15 | IVT4,199 |
| Block-2 | IVT2,11 | IVT3,15 | IVT4,1114 |
| Block-3 | IVT2,12 | IVT3,111 | IVT4,1147 |
| Block-4 | IVT2,11 | IVT3,111 | IVT4,1177 |
| Block-5 | IVT2,12 | IVT3,121 | IVT4,1180 |
| Block-6 | IVT2,12 | IVT3,121 | IVT4,1110 |
| Block-7 | IVT2,12 | IVT3,121 | IVT4,1225 |
| BLOCK | Sequence-1 in 5 adic IVT | Sequence-1 in 6 adic IVT | Sequence-1 in 7 adic IVT |
| Block-1 | IVT5,1194 | IVT6,128565 | IVT7,1297051 |
| Block-2 | IVT5,1214 | IVT6,128595 | IVT7,1297093 |
| Block-3 | IVT5,1294 | IVT6,128745 | IVT7,1297393 |
| Block-4 | IVT5,1334 | IVT6,128805 | IVT7,1297435 |
| Block-5 | IVT5,1414 | IVT6,128985 | IVT7,1297109 |
| Block-6 | IVT5,1434 | IVT6,128955 | IVT7,1297151 |
| Block-7 | IVT5,1694 | IVT6,128960 | IVT7,1297793 |
| BLOCK | Sequence-1 in8 adic IVT | Sequence-1 in 9 adic IVT | |
| Block-1 | IVT8,15135375 | IVT9,1102907844 | |
| Block-2 | IVT8,15135431 | IVT9,1102907916 | |
| Block-3 | IVT8,15135886 | IVT9,1102908572 | |
| Block-4 | IVT8,15135942 | IVT9,1102908644 | |
| Block-5 | IVT8,15138959 | IVT9,1102913676 | |
| Block-6 | IVT8,15139015 | IVT9,1102913748 | |
| Block-7 | IVT8,15139981 | IVT9,1102915132 |
Table 2: IVTs from 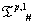 used for generation of
used for generation of
| MDGGNQSEGSEFLLLGMSESPEQQRILFWMFLSMYLVTVVGNVLIILAIS |
| SDSRLHTPVYFFLANLSFTDLFFVTNTIPKMLVNLQSHNKAISYAGCLTQ |
| LYFLVSLVALDNLILAVMAYDRYVAICCPLHYTTAMSPKLCILLLSLCWV |
| LSVLYGLIHTLLMTRVTFCGSRKIHYIFCEMYVLLRMACSNIQINHTVLI |
| ATGCFIFLIPFGFVIISYVLIIRAILRIPSVSKKYKAFSTCASHLGAVSL |
| FYGTLCMVYLKPLHTYSVKDSVATVMYAVVTPMMNPFIYSLRNKDMHGAL |
| GRLLDKHFKRLT |
Text 1: Protein Sequence of OR1D2.
Similarly, other S(ti)can be generated applying the IVTs in different blocks of the S(ti-1)as tabulated in supl.met-I. We have generated 90 such S(ti)s corresponding to OR1D2 in each , system (for p=2, 3… 20) (available in supl. met.-II).
All these generated sequences have been blast in the NCBI database for significant similarity. The blast result is shown in supl. met.-III.
Most of the generated sequences are mapped to olfactory receptors (specifically close to OR1D2) in different organisms like homo sapiens, pan troglodytes, lagothrixlagotricha etc. Some of the sequences are not mapped due to the fact that they are more conserved sequence than OR1D2.
Also we have been observed that some of the protein primary structural properties (listed below) are intact with respect to the two dimensional protein plot graphs (using bioinformatics toolbox of Matlab-R2010b) for each of the generated sequences.
The protein properties which we have considered here are as follows:
• Prop-1: Accessible residues (%)
• Prop-2: Alpha helix (Chou &Fasman)
• Prop-3: Amino acid composition (%)
• Prop-4: Beta sheet (Chou &Fasman)
• Prop-5: Beta turn (Chou &Fasman)
• Prop-6: Coil (Deleage& Roux)
• Prop-7: Hydrophobicity (Aboderin)
• Prop-8: Total beta strand
Corresponding to each property of the S(ti), we have had eight protein plot graphs from which we have calculated box counting dimensions using BENOIT™.
The data for OR1D2 sequence are stated below in the Table 3. The rest of the data are available in the supl. met-IV. We have observed that the box-counting dimensions for all the eight protein plots corresponding to each of the protein property for all the generated sequences S(ti)s are almost same to the same of OR1D2. The data for all the box counting dimension of protein plots for the S(ti) generated through the system is shown below. Hereby we can come to a conclusion that these IVTs preserve the protein properties of the strings. It is to be noted that all these IVTs are bijective; therefore one can switch from one protein to another protein through the IVTs without encumbering the protein properties. Most of the S(ti) (IVT generated sequences) preserve all the eight protein properties. It is to be noted that in the case system, S(t1) and S(t2) are both mapped to G-protein-coupled receptor in OR1D2 in human. Also they follow all the protein properties as in OR1D2 (Table 4).
| Sequence | Property | Box-counting dimension |
|---|---|---|
| OR1D2 | Prop1 | 1.91092 |
| Prop2 | 1.91103 | |
| Prop3 | 1.90855 | |
| Prop4 | 1.91141 | |
| Sequence | Property | Box-counting dimension |
| Prop5 | 1.91095 | |
| Prop6 | 1.91348 | |
| Prop7 | 1.90989 | |
| Prop8 | 1.91071 |
Table 3: Box-counting dimension for protein plots of OR1D2.
| Sequence | Property | Box-counting dimension |
|---|---|---|
| S(t1) | Prop1 | 1.92694 |
| Prop2 | 1.91117 | |
| Prop3 | 1.90976 | |
| Prop4 | 1.91111 | |
| Prop5 | 1.9113 | |
| Prop6 | 1.93038 | |
| Prop7 | 1.91021 | |
| Prop8 | 1.91144 | |
| S(t2) | Prop1 | 1.91124 |
| Prop2 | 1.91099 | |
| Prop3 | 1.91389 | |
| Prop4 | 1.90948 | |
| Prop5 | 1.91064 | |
| Prop6 | 1.93051 | |
| Prop7 | 1.91398 | |
| Prop8 | 1.90983 | |
| S(t3) | Prop1 | 1.91045 |
| Prop2 | 1.91049 | |
| Prop3 | 1.90994 | |
| Prop4 | 1.91299 | |
| Prop5 | 1.92765 | |
| Prop6 | 1.91648 | |
| Prop7 | 1.92813 | |
| Prop8 | 1.91448 | |
| S(t4) | Prop1 | 1.91294 |
| Prop2 | 1.91495 | |
| Prop3 | 1.91084 | |
| Prop4 | 1.9108 | |
| Prop5 | 1.91155 | |
| Prop6 | 1.91577 | |
| Prop7 | 1.9281 | |
| Prop8 | 1.93043 | |
| S(t5) | Prop1 | 1.91443 |
| Prop2 | 1.91431 | |
| Prop3 | 1.91259 | |
| Prop4 | 1.93055 | |
| Prop5 | 1.92909 | |
| Prop6 | 1.91638 | |
| Prop7 | 1.92901 | |
| Prop8 | 1.91676 | |
| S(t6) | Prop1 | 1.92863 |
| Prop2 | 1.928 | |
| Prop3 | 1.91431 | |
| Prop4 | 1.9295 | |
| Prop5 | 1.91133 | |
| Prop6 | 1.91751 | |
| Prop7 | 1.91379 | |
| Prop8 | 1.91292 | |
| S(t7) | Prop1 | 1.91421 |
| Prop2 | 1.928 | |
| Prop3 | 1.9142 | |
| Prop4 | 1.91614 | |
| Prop5 | 1.9101 | |
| Prop6 | 1.91402 | |
| Prop7 | 1.9108 | |
| Prop8 | 1.91314 | |
| S(t8) | Prop1 | 1.9104 |
| Prop2 | 1.91378 | |
| Prop3 | 1.91039 | |
| Prop4 | 1.91287 | |
| Prop5 | 1.91177 | |
| Prop6 | 1.91392 | |
| Prop7 | 1.90987 | |
| Prop8 | 1.91378 | |
| S(t9) | Prop1 | 1.91428 |
| Prop2 | 1.91129 | |
| Prop3 | 1.91367 | |
| Prop4 | 1.91337 | |
| Prop5 | 1.91263 | |
| Prop6 | 1.91431 | |
| Prop7 | 1.91084 | |
| Prop8 | 1.91413 | |
| S(t10) | Prop1 | 1.91082 |
| Prop2 | 1.9108 | |
| Prop3 | 1.91081 | |
| Prop4 | 1.91337 | |
| Prop5 | 1.91263 | |
| Prop6 | 1.91514 | |
| Prop7 | 1.91084 | |
| Prop8 | 1.9176 |
Table 4: Box-counting dimension for all protein plots of in
But interestingly, there are many S(ti ) in different , systems, do not map significantly in any organisms but they possess the protein properties as in OR1D2. One of the main reasons for this is that most of the sequences are conserved whereas OR1D2 is not so. Some of the S(ti ) are not mapped to any of the ORs in any organism although the box-counting dimension for all the protein plots are intact as it is in OR1D2. It is our strong conviction that these S(ti ) serve the purpose for replacement of OR1D2 in the genetic evolutionary future. In the next section we are about to discuss the case on applying the bijective IVTs from 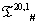 (Table 5).
(Table 5).
| Sequence | Property | Box-counting dimension |
|---|---|---|
| S(t1) | Prop1 | 1.90836 |
| Prop2 | 1.91371 | |
| Prop3 | 1.92937 | |
| Prop4 | 1.91313 | |
| Prop5 | 1.92746 | |
| Prop6 | 1.9128 | |
| Prop7 | 1.91234 | |
| Prop8 | 1.91291 | |
| S(t2) | Prop1 | 1.91418 |
| Prop2 | 1.91204 | |
| Prop3 | 1.91182 | |
| Prop4 | 1.91205 | |
| Prop5 | 1.91418 | |
| Prop6 | 1.92998 | |
| Prop7 | 1.9099 | |
| Prop8 | 1.91351 | |
| S(t3) | Prop1 | 1.91459 |
| Prop2 | 1.91308 | |
| Prop3 | 1.91151 | |
| Prop4 | 1.91464 | |
| Prop5 | 1.91434 | |
| Prop6 | 1.91216 | |
| Prop7 | 1.91306 | |
| Prop8 | 1.91321 | |
| S(t4) | Prop1 | 1.91087 |
| Prop2 | 1.91468 | |
| Prop3 | 1.90957 | |
| Prop4 | 1.90991 | |
| Prop5 | 1.92755 | |
| Prop6 | 1.9159 | |
| Prop7 | 1.9104 | |
| Prop8 | 1.91369 | |
| S(t5) | Prop1 | 1.91448 |
| Prop2 | 1.91485 | |
| Prop3 | 1.92691 | |
| Prop4 | 1.914 | |
| Prop5 | 1.9123 | |
| Prop6 | 1.91203 | |
| Prop7 | 1.92751 | |
| Prop8 | 1.92845 | |
| S(t6) | Prop1 | 1.91315 |
| Prop2 | 1.91176 | |
| Prop3 | 1.91169 | |
| Prop4 | 1.91317 | |
| Prop5 | 1.91348 | |
| Prop6 | 1.91507 | |
| Prop7 | 1.91141 | |
| Prop8 | 1.92879 | |
| S(t7) | Prop1 | 1.91258 |
| Prop2 | 1.91057 | |
| Prop3 | 1.91388 | |
| Prop4 | 1.91508 | |
| Prop5 | 1.92907 | |
| Prop6 | 1.91605 | |
| Prop7 | 1.91244 | |
| Prop8 | 1.91098 | |
| S(t8) | Prop1 | 1.92725 |
| Prop2 | 1.92767 | |
| Prop3 | 1.91331 | |
| Prop4 | 1.91074 | |
| Prop5 | 1.91459 | |
| Prop6 | 1.91608 | |
| Prop7 | 1.90883 | |
| Prop8 | 1.91143 | |
| S(t9) | Prop1 | 1.90984 |
| Prop2 | 1.92917 | |
| Prop3 | 1.9154 | |
| Prop4 | 1.91098 | |
| Prop5 | 1.91336 | |
| Prop6 | 1.91545 | |
| Prop7 | 1.91013 | |
| Prop8 | 1.92845 | |
| S(t10) | Prop1 | 1.91286 |
| Prop2 | 1.91425 | |
| Prop3 | 1.91506 | |
| Prop4 | 1.91402 | |
| Prop5 | 1.92938 | |
| Prop6 | 1.91632 | |
| Prop7 | 1.91337 | |
| Prop8 | 1.9125 |
Table 5: Box-counting dimension for all protein plots of in
On Applying 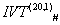 : We have chosen a few bijective IVTs (available in supl. met.-I) from system to generate S(ti) from system to generate S(ti) from the protein code for OR1D2 (methodology is discussed in 3.1). Here all the S(ti) have been blasted in NCBI and they all are mapped to G protein-coupled receptor, or MOR30-1, hypothetical protein and conserved hypothetical protein in different organisms ranging from human to plasmodium species (data shown in supl. met.-III). The box counting dimension is still intact for all the protein plots for all the IVT generated sequence in system as shown in (Figure 1) (raw data shown in supl. met-IV). It is noted that the number of bijective, domain preservative IVTs is increased as p increased in T(p,1)# . Consequently the sequential conservation is inversely proportional to p.
: We have chosen a few bijective IVTs (available in supl. met.-I) from system to generate S(ti) from system to generate S(ti) from the protein code for OR1D2 (methodology is discussed in 3.1). Here all the S(ti) have been blasted in NCBI and they all are mapped to G protein-coupled receptor, or MOR30-1, hypothetical protein and conserved hypothetical protein in different organisms ranging from human to plasmodium species (data shown in supl. met.-III). The box counting dimension is still intact for all the protein plots for all the IVT generated sequence in system as shown in (Figure 1) (raw data shown in supl. met-IV). It is noted that the number of bijective, domain preservative IVTs is increased as p increased in T(p,1)# . Consequently the sequential conservation is inversely proportional to p.

Chart 1: Flow Chart for our methodology for a generalith iteration.
In summary, we have seen that IVTs steer a given OR sequence of a species to another of the same or different (most likely) species, preserving the protein properties of the original sequence. This methodology will be helpful to mimic the genomic evolution procedure artificially, which is required for genetic replacement therapy. IVTs may also be considered to be a platform to comprehend the morphological connections among the various species. A naïve question to the biologists, which rose amongst us:
Suppose, we are given an olfactory receptor or1 of a species s1 which help it to identify the odors x1, x2,…
Now, we apply the proposed methodology to or1 and obtain a new olfactory receptor or2 (supposedly) of species s2.
So, does or2 help s2 in identifying the same odors x1, x2,...?
In the near future, we are really interested to explore the underlying biological methodology that governs the entire process.
Authors would like to thank to their visiting students Anjan Pal and Snehasish Banerjee for their enormous help in writing computer programs.