Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2015) Volume 8, Issue 7
Expression profiling is a popular tool for studying gene expression levels, but libraries’ origins and data quality are often poorly annotated or contain errors. Experimental techniques, library annotations and analysis algorithms vary between laboratories and may contain errors. Traditional analysis methods, including research into tissuespecific expression, assume expression levels to be correct and libraries to be correctly annotated, which is not always the case. Therefore, tools capable of assessing the quality of multiple types of expression data using the data alone would be invaluable for quality control of that data and elucidation of its suitability for expression analysis. Here we compare and review over 20 methods and focus on a number of key developments in the field. We also highlight the application of recently devised novel quality control methods and show examples of applications of the newly developed quality control expression matrixes (QCEM) to the analysis and quality control of SAGE data. The described example include elucidating the correct tissue identity and show that disease state for expression libraries created using a range of expression profiling methods might be easily elucidated. The described novel quality control methods address key shortcomings of the previously reported tools and provide a universal quality control method for multiple types of expression data.
Keywords: Cancer bioinformatics, Differential gene expression, Gene expression profiling, Sample annotation, Tissue-specific expression, Quality control of expression data
mRNA expression profiling is now well established and a number of techniques are employed for acquiring and analysis of data on a sample’s transcriptome or for studying differential gene expression. Some of the most widely used methods include qPCR [1] EST (Expressed Sequence Tags, early 1990s [2]), SAGE (Serial Analysis of Gene Expression) and microarrays (mid 1990s onward [3], and most recently, RNA-Seq [4], to name just a few. Growing amounts of gene expression data have resulted in the growth of large databases such as NCBI’s EST [5], for storing and processing the data and retrieval tools such as those hosted by the Cancer Genome Anatomy Project (CGAP) [6]. However, the abundance of the data and the absence of easily identifiable data quality indicators require stringent quality control methods, e.g. for confirming correct annotation, or the identity of each library independently of the annotation and the quality of the underlying data itself such as bulk non-normalised preparations, or the methods used. Such tools would help to resolve many errors in expression data annotations such as tissue of origin, disease state or protocol used to prepare the library, because even a trivial error in for instance library origin might completely invalidate data selection for the analysis and the results obtained.
Many such errors arise during experimental stages, e.g. cloning artefacts, amplification artefacts and biases, normalisation (intentional or unintentional), the use of different RNA extraction methods, RT-qPCR (Real-Time quantitative PCR) amplification artefacts and shallow depths of sequencing of such libraries, to name a few [7]. Errors can also be introduced from the fact that multiple polyadenylate repeats are found in a significant percentage of mRNA species contain multiple polyadenylation sites, potentially leading to multiple transcripts being produced from one mRNA [8]. While reverse transcription, which is carried out to prepare the sample for amplification, in theory should result in one cDNA molecule for each original mRNA molecule, in practice some mRNA may not undergo the full set of reactions, introducing a bias into the sample in favour of the fully converted cDNAs [7]. The same applies to PCR amplifications, where in practice amplification efficiency of different cDNAs transcripts may differ leading to under- or overrepresentation of a fraction of amplified transcripts [9]. Also, the selection of cDNA clones for sequencing is random, which may introduce biases for low abundance transcripts. Other groups of errors are due to human factors, and such as incorrect data annotation, pooling/contamination of tissue samples, and inadequate depths of sequencing. RNA-Seq, which uses next generation sequencing, provides a much improved depth of sequencing, but artefacts can still be introduced into the results, requiring quality control [10].
To be usable for gene expression analysis, one also needs to ensure the reported tag counts in EST, SAGE, RNA-Seq, and similar other investigations mirror true cDNA abundance levels as closely as possible. For this purpose a large proportion of the library must be sequenced to achieve deep sequencing. While this becomes less of a problem with the advent of RNA-Seq it like all the other methods, will still suffer from “noise” (this includes which are of limited biological significance [11]) introduced during experimental procedures [12-15]. Artificial changes in the results are often introduced by the use of different preparation procedures and kits. For example, various media and kits for RNA extraction, including one kit by Qiagen and another by Promega, were analysed and compared, and the choice of materials had a major effect on the results [16]. Such errors may lead to false positive results or the omission of potential diagnostic biomarkers or therapeutic targets from further investigations, which in turn may lead to erroneous diagnoses or incorrect treatments. Recent developments indicate that quality control measures can be devised for use by the end user who often does not have access to the original raw expression data.
Here we aim to outline the main issues and recently developed quality control methods for testing various types of gene expression data.
There is a range of different gene expression profiling methods available, each of which has its own advantages, disadvantages and the different experimental errors and biases affecting the final results. An old favourite Northern blotting analysis is still in use today; it is carried out by separating a sample of RNA by electrophoresis in agarose according to molecular weight and subsequent transfer of separated RNAs to a nylon membrane. A fluorescent or radiolabelled nucleotide probe complementary to the target gene is then added and hybridised to that gene; the strength of the detected signal will indicate relevant mRNA concentrations. However, with this technique it is only possible to measure the relative abundance levels of a specific transcript of interest, whose sequence must also be previously known for the probe to be produced. Despite this and the low throughput, Northern blotting introduces the fewest errors or biases into the data when undertaken using more recent protocols [17] (Figure 1). Unfortunately, the limitations of camera detection systems (typically the limited dynamic range or limited sensitivity, respectively) and some experimental artefacts such as RNase contamination, may still lead to some normalisation of the results [17]. A detailed discussion of these and other problems is available from [17,18].

Figure 1:Gene expression profiling methods and the differences between them. The raw data output format is shown for each of the six methods. Reverse transcription is not required for the two methods on the left (northern blotting and microarrays), both of which are performed directly on mRNA. Reverse transcription is required for the other four methods. The initial tissue sample can be from any source tissue or organism and can be prepared in a variety of ways (bulk tissue, cell line, etc). Three of the methods (RNA-Seq, ESTs and SAGE) utilise absolute tag counts and are therefore referred to as “digital” methods, while northern blotting, microarrays and RT-qPCR are referred to as “analogue” because they measure relative abundance levels (although they can be made absolute through use of an internal standard). The table below the figure summarises individual features of relevant techniques, provides a qualitative comparison of common expression profiling methods and compares the likelihood of errors entering the results. Detailed analysis of the individual methods are beyond the scope of this paper and we refer readers to original publications and recent reviews of these. See e.g. [138-139] for Northern blotting, [140-141] for microarrays, [142-143] for RNA-SEQ, [144-148] for PCR based methods, [149-150] for EST profiling and [151-152] for SAGE profiling methods.
Real time quantitative RT-PCR is widely used for measuring mRNA levels; it offers unsurpassed sensitivity and moderate throughput, for a detailed review see [1]. This technique provides information on the relative differences in the abundance of one or more genes of interest in different samples after normalisation using e.g. ribosomal RNAs as an internal standard. Real time PCR is advantageous compared to Northern blotting because of the superior sensitivity and throughput, but the threshold at which sample fluorescence is deemed significant compared to background is open to interpretation and is not objective.
Furthermore, if the original mRNA is fragmented (as it is likely to be from archived samples), use of oligo(dT) as a more specific primer to ensure more faithful replication, is less effective [1]. In addition to being prone to errors, this method is also less quantitative due to its reliance on standard curves for absolute abundance levels. The major source of bias for RT- qPCR comes from variations in efficiency of both reverse transcription and the PCR amplification itself, which is often far from ideal [9], introducing biases into the results towards transcripts which are amplified more than others. As a result of chemical kinetics, these will often be the transcripts which were more abundant originally. Furthermore, long transcripts and/or those which are GC rich, can be amplified less efficiently than the other transcripts, due to the increased melting point of such transcripts [18]. This results in a GC content bias, which also occurs in EST and SAGE (see below). These sources of error are often addressed by using an internal standard to obtain absolute quantitation. A standard is amplified simultaneously with the target and all other values are normalised against it [19].
RNA-Seq uses next generation sequencing equipment often that produced by Illumina or Roche [20]. An RNA population is converted to a library of cDNA fragments which have adapters ligated to at least one end. The library is amplified if necessary before sequencing from one or both ends. The fragments and reads are usually in the order of a few hundred base pairs in length [21] and are mapped onto the original cDNA sequence (Figure 1).The high depth of sequencing eliminates the need for normalisation [21] for novel gene discovery. However, the results cannot be deemed reliable if used for gene expression analysis, without much replication, standardisation and calibration, for each protocol has its own biases [22] RNA-Seq requires cDNAs to be fragmented, introducing a bias towards fragments taken from the 3’ end of transcripts, with each fragmentation method introducing its own bias. If amplification is required this could introduce artefacts in the form of many identical short reads [21]. This is because factors such as GC content or mononucleotide repeats which introduces biases into the data are largely consistent between lanes of the same run, or between multiple runs of the same cDNA concentration [4]. The GC content or the abundance of these repeats can be measured to give an indication of experimental biases or defects introduced during library preparation [23].
DNA microarrays and similar chip-based methods containing immobilised nucleotide probes, provide excellent coverage and throughput, and usually require prior knowledge of all of the target sequences. Differential expression is inferred from relative differences in the fluorescence readings which result from differing degrees of hybridisation of those probes (oligonucleotides, cDNAs, other DNA fragments) with different samples of interest which are applied to the chip [24]. Pioneered by Affymetrix [25,26], oligonucleotide microarrays are chips containing as many as 1.3 million [27] oligonucleotide sequences, enabling greater coverage of the transcriptome. The key advantage of microarrays over RT-PCR is the extent of coverage, often allowing many more genes to be studied in one experiment [28,29] (Figure 1). Unlike real time PCR, microarray based methods measure differential gene expression which is inferred from changes in relative fluorescence levels between samples whilst absolute levels of gene expression are not determined. However a great deal of replication, standardisation (of protocols and reporting methods [30] and calibration is required for accurate comparison of expression patterns between different arrays or when comparing different sources or individual experiments on one sample in which different types of chip are used (this particularly applies to cDNA microarrays). Furthermore, signal intensity can be affected by the length of the immobilised nucleotide probes as well as their composition (molecules of more than one molecular species in the sample are more likely to hybridise with template molecules of one species if that template contains large numbers of repeats) and the methods used for generating labelled probes [31]. In order to account for these sources of error, replicates are normally performed from the same sample, all of which are then normalised to reduce the variation between chips [32]. Furthermore, the lack of sensitivity which results from a gene being reported as only upregulated or downregulated can be addressed through statistical methods such as Principal Component Analysis to retrieve the useful data from the noise [33].
Apart from RNA-Seq, the above methods are considered “analogue” because they involve the measurement of relative abundance levels. RNA-Seq may be considered “digital” because it relies on the tag counts. Another “digital” methods in use relies on counting ESTs (Expressed Sequence Tags) or Serial Analysis of Gene Expression (SAGE). Having been developed in the early 1990s, this approach was originally used for novel gene discovery and gene mapping [34-36], but the ever-increasing amount of data has subsequently allowed EST data to be used to investigate gene expression levels and to study differential expression between different tissues or conditions. EST expression data are available from NCBI [5]. Some specialised databases e.g. CGAP allow user to compare EST expression levels in cancer and normal tissue in silico [36]. Traditionally, EST libraries are created by sequencing randomly selected transcripts in a cDNA library [37]. These are then assembled into longer, overlapping sequences mapped onto the original transcript and a unique UniGene Cluster ID is assigned to each transcript. mRNA expression levels are inferred by counting the absolute number of tags representing each transcript. EST libraries therefore contain a snapshot of mRNAs expressed in the sample from which the library was created [38]. However, the sensitivity is often reduced compared to northern blotting and real time quantitative RT-PCR because the depth of sequencing is lower than with RNA-Seq (Figure 1). EST based approached were first developed in the 1990s [39] and are still used today and the total EST counts often exceed 10,000 per library.
Because the purpose of an EST libraries was to assist gene discovery rather than to study expression profiling, the EST content of a library was often altered to reduce the abundance of transcripts representing genes with high expression. To achieve this, EST libraries were normalised or subtracted by removing the most abundant or less relevant transcripts in order to reduce or eliminate the differences in the relative transcript abundances to a narrow range [40-43]. However, any normalisation introduces a major bias towards rare transcripts, so such libraries are not normally suitable for a fully quantitative gene expression profiling. Once a cDNA library has been created, ESTs are produced by sequencing randomly selected cDNAs from a library, usually from the 3’ end to generate single read fragments which are often longer than several hundred base pairs in length. However, the sequencing can be performed in a number of different ways, leading to, using the CGAP database as an example, three different groups of libraries (CGAP, MGC and ORESTES). ORESTES libraries are generated differently from the other two by being sequenced from arbitrary points in the middle of each cDNA instead of either end [37]. These different methods have a multitude of biases and may produce different results. Furthermore, smaller EST libraries miss rare transcripts due to shallow sequencing [44]; a greater depth of sequencing is required for a better quantitative estimate of gene expression in the original sample [45]. Therefore, while EST based methods provide wide gene coverage, the sensitivity is often low due to the low depth of sequencing of some libraries. Alarmingly, the existing algorithms used to analyse EST expression data place the emphasis on identification of the degree of over/under- expressed for tissue/disease-specific genes without fully evaluating the quality of the expression data or the origins of the experimental material used to produce EST libraries.
A technique called Serial Analysis of Gene Expression (SAGE), also involves the production of tags by sequencing a cDNA library [46]. However, unlike ESTs, each SAGE tag is a short transcriptspecific sequence of 9 – 26 base pairs in length, and many such tags are concatenated together into one cDNA molecule prior to sequencing, which improves throughput and the sequencing depth and coverage compared to the EST approach. SAGE tagging allowing at least in principle 49(i.e. over 262,144) potentially unique sequences. This makes SAGE another “digital” high-throughput method of gene expression profiling. SAGE requires fewer sequencing runs than ESTs for a representative profile of gene expression levels to be produced, leading to greater quantitative analysis and a reduction in the introduction of errors or biases compared to ESTs (Figure 1). The disadvantage of SAGE compared to ESTs is that while the tag length of 9 – 26 base pairs is in theory sufficient to identify all transcripts in any mRNA sample, in practice individual SAGE tags often map onto multiple transcripts [47-54]. For example, out of 130,029 different transcripts in UniGene database (as of 13November 2012), in the Cancer Genome Anatomy Project Short SAGE Database of 1,048,576 tags, 321,674 mapped onto a UniGene Cluster, and of these, only 64,412 had one tag sequence uniquely mapping onto them [55]. It is because of this problem of ambiguity, that quality control is required. In addition to the problems stated above for ESTs, the shorter tag length increases the risk of ambiguity in the results [31,32,55,56]. However, due to the concatenation, the depth of sequencing is greater with SAGE so the sensitivity will be higher. Sequencing errors can still arise, and estimates of such errors can be generated. Statistical algorithms have been created to correct for these errors [57], but no all-inclusive solution to these problems have yet been reported for SAGE expression data. An ideal method should be able to indicate the degree of normalisation of a normalised library or for cancer staging.
A few studies have been performed into quality control for most of the methods discussed in this article. In one investigation involving SAGE data [58] three databases were compared – Gene Expression Atlas (oligonucleotide microarray data), SAGE map (SAGE libraries) and Tissue Info (EST libraries). Because these databases use different formats for sample annotation and use different statistical methods for data analysis, a method called Preferential Expression Measure (PEM) was devised to score differential expression of genes in libraries grouped into six different tissue categories (brain, kidney, ovary, pancreas, prostate and vascular endothelium) in three databases. Interdatabase correlations were measured and were found to be high for brain, prostate and vascular endothelium, but not for kidney, ovary and pancreas. However, inter-library correlations have yet to be applied as a quality control method within one database [58]. However, the invention of PEM shows that quality control of data between databases has been attempted and should be explored further while also working well for all tissues within a single database.
In another study, data for 8,570 genes across 46 human tissues from the Gene Expression Omnibus (an Affymetrix microarray data repository) were categorised according to tissue specificity and subcellular localisation of their protein product [59]. The authors reported that widely expressed genes have higher expression levels than genes which are expressed in one or a few tissues [59]. However, only a third of the available genes were ever included in the study, which means that it would not be possible to utilise this as a quality control method for annotating new samples or accurately characterising existing less well characterised samples.
In a report by Daley and Smith [60] the problems related to shallow sequencing were considered in the context of obtaining sufficient gene coverage. A statistical method was proposed to provide an indication of the depth of sequencing required to completely sequence a library from an initial shallow sequenced sample. This improved on previously tested methods where problems arose when attempts were made to extrapolate beyond twice the initial run, leading to incorrect predictions. The new method was found to correctly predict when saturation would or wouldn’t occur [60]. However, the reported method, which utilised Bayesian statistics, did not take into account experimental errors or biases introduced during procedures prior to sequencing.
The problem of obtaining sufficient gene coverage was addressed successfully by the introduction of RNA-Seq, which provides a substantially greater depth of sequencing compared e.g. to EST. However, this method is still plagued by artefacts, requiring quality control to be applied. This has so far extended to the detection of poor quality sequence reads [10], which while useful, would not detect, for example, contamination of one tissue with another or the degree to which gene expression in cancer has diverged from that of the primary tumour’s host tissue, which will be significantly greater in a secondary metastasis (which may be mislabelled as originating from the primary tumour [61]).
As with “digital” methods such as RNA-Seq, “noise” can be introduced into oligonucleotide microarrays (also known as “tiling arrays”) during steps such as RNA isolation and handling, cDNA amplification, labelling and hybridisation, and during scanning of the microarrays. Various attempts have been made to indicate the statistical significance of results in order to discern signal from this socalled technical noise [12-15]. However, correction of “noise” will not solve problems of sample contamination and mislabelling, which will go undetected because tissue-specificity of expression is not taken into account.
Attempts made to improve the quality of samples and to control for tissue-specificity can be exemplified by the investigations into the effect of RNA degradation post-mortem [62,63]. Fixing protocols may prevent mRNA decay but have a potential to introduce their own artefacts into the results [64,65]. Therefore a quality control test is required that can be used to tissue type a sample independently of external information. Krzystanek et al. [66] devised the “Biasogram” statistical approach for this reason, but this method does not address other pitfalls such as source material quality or cross-tissue contaminations, which a quality control method needs to in order for it to be useful in elucidating the tissue origin of a sample. Methods have also been devised to quality control transcript annotations as they are applied to microarray data, and these have been included into tools built for transcript annotation [67,68]. However, even if the transcripts are correctly annotated, this will not be enough to verify correct annotation of the tissue sample as a whole. While methods have recently been devised for gene expression profiling of a single cell [69], “noise” can still be a problem, and while methods have been devised to address this [70], results from studies of a single cell will still be incorrectly processed and subsequently studied erroneously if the tissue sample from which that cell was derived was wrongly annotated.
Other investigations have compared the results from different gene expression profiling methods when applied to the same sample, which is important as all the methods are vulnerable to errors or biases to some degree. For example, Malone and Oliver [71] compared the results obtained from RNA-Seq and microarrays in a study of gene expression between the male and female heads of the fly Drosophila pseudoobscura. They found the results were the same, but they did not use this to create a quality control method which could be used to validate the identity of samples. Thus sample contamination would remain undetected. Furthermore, this investigation was carried out on non-clinical samples, a problem rectified by Turnbull et al when they compared microarray and RNA-Seq data from breast cancer samples to assess whether it was possible to combine the two types of data for further analysis. Though this was found to be possible, it was not extended to other tissues, which would have been a prerequisite for any attempt to use it as a quality control method for sample tissue typing [72]. An opportunity to devise a method for quality control of sample annotations was also missed in an attempt to devise software for comparing experiments performed using different microarray platforms, for the annotations were all entered manually [73], as they were for INMEX, software designed to integrate transcriptomic and metabolomic data, although the latter did provide a method of verifying the consistency of molecular annotation. However, the algorithm did not include sample annotation verification [74]. Conversely, storage and retrieval of sample metadata, which includes tissue annotation, was the focus of a data management package called the eGenVar. Whilst it includes scripts for automatically adding the data for many samples at once, scripts were not included to verify the annotation based on the associated raw data, and the system was only capable of handling microarray data [75].
Kadota and Shimizu [76] came close by using groups of genes instead of single genes to infer differential gene expression, and they ranked the genes for this purpose, but they did not extend this gene set enrichment to quality control of sample annotation, allowing sample contamination or incorrect annotation to remain undetected and unresolved. Similarly, Wen et al. [77] used genes which were common detected by different microarray methods as a quality control method for comparing the methods, but this was not extended to using a defined set of tissue-specific genes to quality control the sample annotations. This opportunity was also missed by Zhang et al. [78] when they compared the results of statistical analysis of gene expression data obtained from a range of microarray platforms. The opportunity to study tissue-specific expression in such a comparison was taken by Luo et al. [79] but the annotations were assumed to be correct and a quality control method to verify this was not devised.
While quality control methods were previously suggested, they only focussed on the genome (potentially missing alternatively spliced variants that would have been detected by studying the transcriptome) [80,81], or on the proteome (potentially missing mRNAs whose translation is downregulated by microRNAs [83,84] or covered aspects of the data such as GC content [40], noise [12,13] source material quality [62,63,67,70,86], different experimental methods [64-66,71-74,76-79] or read quality [87-95], with few investigations focusing on the tissuespecificity issues [75,96], even when two or more methods were used in the same study [97-101]. A common shortcoming of many previous attempts is that tissue specificity of the genes was reported [102-112], or avoided [113-115]. However, no attempts were made to actually use such data for quality control or evaluation of the expression data, or if they were, it was for cancer analysis within one tissue [116-118] or to study the expression of synonymous codons in plants [119]. This is despite the fact that questions over the validity of gene expression profiling results in breast cancer, for example, have been discussed [120]. Even where an attempt has been made to summarise gene expression data, it was only applied to microarrays [121,122] and not transferred to other types of expression data. What is also needed is a method which, once devised, does not require adjustment of sample data for it to work, as has been the case previously [123-125]. Moreover, even unique “tissue specific genes” might be of little practical use if they are expressed at low levels and would therefore be absent in many smaller libraries or not detected in smaller size samples, after all, the general tendency is to miniaturise the assays and samples with the ultimate goal of single cell analysis [126-128]. A greater depth of sequencing would provide a better quantitative estimate of gene expression [48] because lowabundance transcripts are more likely to be included [44], making the library more representative of gene expression in the original sample. However, a simple extrapolation from shallow test runs may not always indicate the true library complexity. The effect of library size on gene expression results has been previously studied and/or taken [cancer [129-135]. However, despite it being known that library size affects the reliability of the results, no comprehensive investigation into quality control has been reported [136].
The amount of effort spent generating and validating expression data is vast but there remain a few shortcomings as discussed above. Recently a new quality control method based on tissue-specific gene expression has been reported [61]. That method does not reply on individual so called “tissue specific” genes; instead the identity of the sample is based on the overall similarity of gene expression patterns. The original quality control expression matrix (QCEM) was developed and validate using human EST expression data. Multiple stages of selecting suitable candidates for inclusion into the QC matrix were: (i) identification of large groups of genes which have similar expression patterns, (ii) prioritising for higher abundance transcripts (for the ease of detection) and (iii) prioritising for groups of genes with highest variability of expression between tissues and organs and lower variability of expression within the same tissues. Following original selection the QCEM subsets were further optimised by selecting genes having lowest correlations of their relative expression levels between different tissues and then by selecting for the highest intra-tissue correlations of their relative expression levels. The selection procedures did not discriminate for age or gender, the training set represented male and female tissues equally and included expression data form different age groups. The QCEM was designed to discriminate and confirm tissue origins not age groups or gender. The development of that approach is described in [61] and the expression matrix is freely available on-line (see e.g. Supplementary Dataset S4 (http://journals.plos.org/plosone/article/asset?unique&id=info:doi/10.1371/journal. pone.0032966.s005) from [61]). In summary the QCEM method relies on the tissue specific ratios of expression of the optimised subset of selected human genes, rather than on the presence of or measuring expression levels of the so called tissue specific genes. The use of expression ratios increases resolution and multidimensionality of the method. Unlike other similar methods based on the “intrinsic” gene subsets, utilising the expression ratios allows distinguishing tissue origins using smaller gene panels. The optimised dataset was dubbed quality control expression matrix (QCEM) and original studies showed it to be able to identify tissue origins for human EST expression libraries and to identify uncharacterised or un-annotated libraries.
The use of QCEM approach requires calculating Pearson productmoment correlation coefficients using expression values from the experimental library of interest with each of the entries in the QCEM:
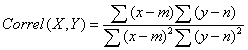
Where x and y are total EST counts for the selected transcripts concerned in QCEM (X) and experimental library (Y0, m and n are the mean EST counts across all transcripts in QCEM control tissue X and experimental library Y [61,137]. The key advantages of using Pearson’s coefficients are that it reveals a degree of dependence between the standard and experimental datasets (QCEM and unknown library respectively) and is independent on the scale of the two variables. I.e. the expression data may of different sort may be used and many linear data transformations are allowed and will not change the outcome of the analysis. Therefore this method is especially applicable for comparing gene expression data generated using different techniques. A library is considered to be a good tissue-specific match if it shows high positive correlation with one specific tissue (and perhaps lower correlation but still significant with a functionally related tissue) and correlation values of close to zero with the other tissues. The use of this approach allowed identification of the tissue of origin as well as distant but related tissue types of human EST expression libraries [61]. Figure 2 shows a few examples of such libraries with correct tissue identity. The robustness of the approach was also confirmed by assessing the degree of normalisation of expression libraries by testing the matrix against EST libraries annotated as normalised as well as with two model normalised libraries in silico. The approach was also tested against randomised libraries as well as cancer EST libraries to attempt cancer staging studies [61]. We subsequently applied the same QCEM for the quality control and identification of small libraries [137]. Although the method was originally developed for use with EST expression data, the generated expression matrixes and the calculations (Pearson productmoment correlation) should be usable with other type of expression data such as for example SAGE, RNA-Seq, microarrays or any other similar methods. To illustrate this capability and as the next step towards establishing a universal quality control method applicable to all forms of expression data here we report the application of the original QCEM to the analysis and quality control of SAGE expression data.

Figure 2:Correlation of the matrix with EST libraries from tissues showing no inter- tissue correlation. Pearson product-moment correlation coefficients (vertical axes) calculated for each individual EST library and the EST expression matrix. A: Placental libraries. B: Lung libraries. C: Retinal libraries. All libraries show high correlation scores with the expected tissues of origin indicating correct library annotations and good expression data quality. Reproduced from [61] with permission.
Here for the first time we apply the same QC matrix (EST derived) to identify and characterise SAGE libraries. Figure 3 shows the results for SAGE libraries from the same tissues as shown in Figure 2, in which the results for EST libraries are presented. While there were only 377 SAGE libraries available in total (as of 9 March 2013) compared to a total of 8,907 EST libraries (as of 25 February 2012), the relevant SAGE libraries provide very high

Figure 3:Correlation of the matrix with SAGE libraries from tissues showing no inter- tissue correlation. Pearson product-moment correlation coefficients (vertical axes) calculated between each individual SAGE library and the QC expression matrix, showing a high quality match for all SAGE libraries. The intra-tissue correlation (with the stated tissue) and inter-tissue correlation (with other tissues) reveal the quality of the match between the two data types. All libraries show high correlation scores with the expected tissues of origin indicating correct library annotations and good expression data quality. The layout is identical to that shown in Figure 2. The result demonstrate that our QC matrix originally created based on EST expression performs equally well when allied to SAGE expression data.
quality matches with their annotated tissue of origin. The sampled SAGE libraries represented a range of ages from foetal to 88 year old tisue and male and female tissues were represented equally. QCEM showed no discrimination, as expected, proving the robustness of the approach. The result shows that the QCEM can be used for tissue typing and characterisation of SAGE libraries as well as EST libraries, showing its potential as a quality control method for multiple data types. This is also an improvement of the earlier reported attempts to quality control multiple data types for a small group of tissues [58]. We further decided to apply the QC matrix to the identification of less well characterised or annotated SAGE libraries. A small number of libraries annotated as being from tissues other than those represented in the matrix and therefore identifiable were used.
Figure 4A shows the tissue identity of four uncharacterised libraries. One such library, annotated as originating from peritoneum (SAGE_ Peritoneum_normal_B_13). While the correlation with the peripheral nervous system would be expected because the PNS is systemic, the correlation with the heart and muscle suggests that the samples were probably contaminated with the related pleura, the serosa which lines the thoracic wall rather than the abdominopelvic cavity. Therefore using this library should not be used for expression profiling studies would yield erroneous results. Another example is the white blood cell library (SAGE_Leukocytes_normal_B_1), which shows strongest correlation with colon and vascular tissues, probably indicating that this library was derived form from blood, but contaminated with colon tissue. The above examples show the robustness of our approach in elucidating the suitability of libraries for expression profiling.

Figure 4:Correlation of the QC matrix with SAGE libraries from uncharacterised or cancer libraries. Pearson correlation coefficients (vertical axes) calculated between individual SAGE libraries and the EST-derived QC expression matrix. The selected examples illustrate uncharacterised, incorrectly defined or cancer libraries indicated by their correlations which contradict libraries’ annotations. A: uncharacterised or ill-defined preparations (left to right): peritoneum library SAGE_Peritoneum_normal_B_13 (expected match: liver, gastrointestinal tract and colon), white blood cell library SAGE_Leukocytes_normal_B_1 (expected match: vascular), “SAGE_Esophagus_Normal_B_CN01” library (expected match: gastrointestinal tract) and “SAGE_GallBladder_Normal_B_HN” expression library (expected match: liver). B: cancer libraries (left to right): brain cancer library “SAGE_Brain_medulloblastoma_B_98-05-P608”, brain library “SAGE_Brain_ glioblastoma_B_H1110”, brain cancer library “SAGE_Brain_astrocytoma_grade_I_B_H1043”and brain cancer library “SAGE_Brain_astrocytoma_grade_III_B_ H1020”.
The usefulness of this method for quality control was further confirmed from the results obtained from a library annotated as originating from oesophagus (SAGE_Esophagus_Normal_B_CN01). While there is slight positive correlation with colon, to which oesophagus is related, this library correlates strongly with brain, skin and with breast, suggesting that it is probably derived from mixed tissue preparations or was normalised and is therefore unsuitable for use in gene expression studies. Furthermore, the final library in Figure 4A (SAGE_GallBladder_Normal_B_HN) is annotated as originating from the gall bladder and the ventral body wall. However, the highest positive correlation is with the nearby colon, strongly suggesting contamination with this tissue. While both tissues would be highly vascularised, resulting in the correlation with vascular tissue, correlation with the thyroid would not be expected if the portion of the ventral body wall collected surrounded the abdominopelvic cavity, which contains the colon and gall bladder. The likely explanation would be that a portion of the wall surrounding the thoracic cavity was collected instead, resulting in the contamination with the nearby thyroid. Because it is a mixed tissue library, this library is more suited to gene discovery investigations than gene expression profiling studies. These examples show that the matrix can be used to verify the identity of un-characterised SAGE libraries, reveal annotation or experimental errors and elucidated whether they are suitable for expression profiling, as was also the case with EST libraries [61].
We also investigated the potential use of the QC matrix as a means for cancer staging. While not enough SAGE data was available to illustrate detailed staging, we were able to elucidate whether such libraries were correctly annotated. Figure 4B shows a few random examples of cancer libraries. “SAGE_Brain_medulloblastoma” is a brain library which correlates well with the stated tissue of origin, suggesting to be correctly annotated. The brain library “SAGE_ Brain_glioblastoma_B_H1110” correlates with peripheral nervous system instead of brain. While this is possible because of the close relation between the two tissues, it may also be incorrectly annotated. Another library (SAGE_Brain_astrocytoma_grade_I_B_H1043) is also annotated as being derived from brain, but it shows clear correlation with liver, suggesting either annotation error or sample preparation error or that it was in fact taken from a secondary metastasis in the liver. The final library presented in Figure 4B is (SAGE_Brain_astrocytoma_ grade_III_B_H1020), also from brain. This appears to be incorrectly annotated, possibly as a result of lacking any differentiation, which does occur in later stages of cancer.
Expression profiling remains a popular approach for studying gene expression levels, but data origins and quality are often not adequately described and may be of inferior quality. Experimental techniques, library annotations and analysis algorithms vary between laboratories and may contain errors. Traditional analysis methods, including research into tissue-specific expression, assume expression levels to be correct and libraries to be correctly annotated, which is not always the case. Multiple tools capable of assessing the quality of multiple types of expression data have been reported but few if any rely on the expression data alone .i.e. on the internal controls, for quality control and suitability of the data for gene expression analysis. Among such tools, the OCEM approach to the tissue-specificity and quality control issues [61,137] showed greater promise. It is different from the previously reported tools in that the origins of the expression data were looked into and the tissue specificity of the original preparations and the data quality are both assessed. The QCEM expression matrix can be used to confirm tissue identities of EST expression datasets for all main human tissue types (Figure 2 for three example tissues), to provide insight into the origin of uncharacterised libraries, to identify normalised or subtracted libraries or various other experimental artefacts. In a few cases it was possible to identify the location of the tumour from which a cancer sample was taken, an extension not previously considered and not previously reported [61]. Furthermore, this approach could be used to correctly identify very small libraries [137], which will have a lower depth of sequencing and will therefore not provide as good a quantitative estimate of gene expression than larger libraries [45] due to the reduced likelihood of rare transcripts being included [44]. The effect of library size has been included previously in statistical tests, which have been used to study gene expression levels in a range of cancers [129-135], but inter-library correlations were not (unlike QCEM, where these were also considered). When applied to SAGE expression data, the correlations illustrated in Figure 3 revealed that QCEM matrix is even more versatile than it was originally thought [61], for it was possible to identify and characterize SAGE libraries as accurately as EST libraries (Figure 2).
The results for the uncharacterized tissue libraries presented in Figure 4A further confirm the potential of the QC matrix as a means to elucidate the origin of libraries whose identity is unknown or not annotated in the database record. It was possible to identify the tissue origin of all four libraries, which in all cases showed contamination with another tissue. Therefore, these findings show the matrix can be used to identify incorrect annotations using both EST and SAGE data as well as verify the identity of libraries that have been correctly annotated.
Cancer libraries are known to show changes in transcription which are characteristic of the type of cancer from which they were taken. As the disease progresses, gene expression is known to increasingly cease to resemble normal gene expression in the tissue where the primary tumour arose. When we came to apply our method to SAGE cancer data, as we had for EST [61], we found that the results gave an indication of the stage of the disease for both types of data, suggesting its potential for indicating cancer progression more accurately than from annotations alone.
We envisage that this approach may be adapted and applied to other expression data such as from DNA microarrays, RNA-Seq data, RT-qPCR and Northern blots. We believe that increasing amount of available data could further decrease the number of transcripts in the expression matrix and may allow accurate analysis and tissue typing of the related and dependent tissues. Merging of this data could bring further improvements, for the quality control method would only need to be assessed once, rather than testing it on each type of data.
The ability to apply a quality control method to the existing gene expression data would be invaluable because expression profiling methods and different procedures used for those methods lead to a range of errors or biases being introduced, different for each procedure or method. This would quality control and correction of data for not only cancer, but also infectious diseases, where gene expression is involved in the regulation of immune responses, and many other disorders, leading to better application of diagnostic or prognostic techniques and novel treatments [138-152].
Expression profiling algorithms were previously found to contain errors, correction of which would ensure the results from investigations into differential gene expression are no longer affected by such problems. However, the results are still dependent on the gene expression data itself being correct, which existing algorithms assume to be the case. While many investigations have previously been undertaken towards quality control of gene expression data, none of them focused on sample tissue type annotation, and no attempts to devise a method to verify this were reported. In other studies where tissue-specific expression was investigated or focused on, no use of this as a quality control method to verify tissue type annotations was presented.
It was previously shown that the tissue type annotations of EST libraries could be verified independently using an expression matrix based on tissue specific markers, showing this to be a suitable means of quality control, which could also be used for cancer staging of EST data. Furthermore, the robustness of the new method was confirmed by using it to correctly identify libraries containing only a handful of ESTs. Here we applied the QC matrix to SAGE data and found it to be equally capable of verifying the tissue identity of SAGE libraries and also for cancer staging. Together, these findings increase the reliability of differential gene expression investigation results for cancer, eliminating the possibility of such errors leading to misdiagnosis, erroneous prognosis or incorrect administration of therapy.
This research was supported by Royal Holloway, University of London. There is no conflict of interest.
ATM carried out the experimental work and contributed to the drafting of the manuscript. MS conceived and supervised the study, and drafted the manuscript. Both authors read and approved the final manuscript.