Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2011) Volume 4, Issue 8
[GADV]-protein world hypothesis [1] leaded me to quantification of decapeptides assembled from G, A, V, D in human proteome. The G, A, V, and D amino acids were related to the nucleotides Guanine (g), Cystosine (c), Uracil (u), and Adenine (a). The search revealed agreement with the genetic code. The types of prebiotic peptide bonds represent probably the first selection power that established the base order in the codons. The genetic code underwent three phases of formation, which explain why modern codons have their particular order of nucleotides: the monobase, dibase and the modern phase (tribase). Sequence alignments and 3D structures of aminoacyl-tRNA synthetases confirm the depicted picture of “relatedness” and the picture indicates how “relatedness” is used by aminoacyl-tRNA synthetases for navigation into and within of the C-terminal anticodon-binding domain. The findings presented here illustrate the novel concept of possible translation of the amino acid sequence into a nucleotide sequence that can be in interactive or contrary mode regarding to desired protein-RNA interactions. Hopefully, it could be used in synthetic biology.
Keywords: Origin of genetic code, Peptide bond types, Human proteome, Nucleotide order of codons, Protein-RNA interactions, Aminoacyl-tRNA synthetases.
It seems that determining the origin of the genetic code is a much more challenging issue then deciphering the code. The state-of-the-art for the origin of the code still remains one of the fundamental unsolved problems. Generally, five basic alternatives exist:
I. The protein world in the beginning - protein coacervates or protein microspheres have developed proto-cells and the genetic code [2-4].
II. The RNA world in the beginning – polymerization of amino acids by poly-RNA [5,6].
III. A parallel evolution of the genetic code and protein synthesis [7].
IV. Transformation of other life systems based on the conditions on the Earth [8].
V. The genetic code and life has been designed by intelligence.
Alternatives IV and V are not within the scope of this manuscript; therefore we will only address the first three. Proteins (I) represented the first target for scientific exploration; however, because RNA (II) was shown to act simultaneously as an informational and catalytic molecule [9], it became the most accepted theory. The scenario in which the genetic code and protein synthesis evolved in parallel (III) resulted as a combination of the two preceding theories. The RNA world theory postulates that self-replicable RNAs (ribozymes) evolved from primordial soup, independently of proteins, and their cellularisation created the first proto-cell, from which followed the evolution of transcription and then translation [6]. Therefore, phylogenetic rooting of transfer-RNAs is a powerful tool that is used to study the early evolution of life and the emergence of the genetic code. For example, Sun and Caetano-Anollés have built phylogenies derived from the sequence and structure of tRNAs and generated timelines of amino acid charging and codon discovery [10]. This rooting showed that charging of Sec, T, S, and L appeared to be ancient, whereas specificities for Q, M, and R amino acids were derived. However, codons for A and P were identified as the most ancient according to their detection. Their study indicates the separate discoveries of amino acid encoding and charging (the genetic and operational code of tRNA). An analysis of a model situation where early tRNAs were not selectively charged with amino acids revealed that it is possible to observe a coded polymerization [11]. Considering this system, a coding regime could have naturally occurred primarily under prebiotic conditions and the operational code could have evolved secondarily [11].
RNA world theory seems be correct; however, the development of amino acids-DNA affinity chromatography [12] over the last decade has revealed that amino acid-nucleotide biomolecular recognition is realistically observed, and it is difficult to imagine any stage of evolution without the influence of peptides. Shimizu showed that single amino acids are able to act as catalysts and anticodonic tri-ribonucleotides corresponding to the amino acid (for example, “uuu” to lysine) act similarly in specific metabolic reaction [13-15]. The idea that amino acids are specifically “related” to nucleotides is not new. Woese probably first suggested the relationship between all amino acids and their codons [16]. He proposed the existence of a “codon-amino acid” logic at some earlier stage in evolution, where he describes amino acidnucleotide interactions as follows:
“It will not do to refer to the usual “picture” of a molecule garnered from 2-dimensional formulas or their 3-dimensional equivalents, for this purpose, because these give a picture of only one type of interaction of which a molecule is capable (i.e., van der Waals repulsion). “Relatedness” should be defined in terms of a composite of all the interactions of which a molecule is capable and these in a proportion defined by the context in question.” [16].
A BLAST search of human taxid: 9606 is used here as the ideal marker of “relatedness”, which was not possible at that time because bioinformatics technology had not yet been developed. The search is based on the new idea that the RNA bond formation was driven by peptide bond between two amino acids with each interaction being specific for a nucleotide. The human proteome is chosen for search because it is only one organism and the genome is completely sequenced. Sequences of most concentrated peptides are used for the nucleotide pairing.
Aliberti thought that the affinity of different amino acids to bind to specific nucleic acid triplets is insufficient and he alternatively proposed the idea of repetitive interactions between specific amino acid side chains and specific bases and between polypeptides and polynucleotides [7], which supports the theory of the parallel evolution of the genetic code and protein synthesis (III). CLUSTAL W Multiple Sequence Alignments of aminoacyl-tRNA synthetase (aaRSs) anticodon loops, and the crystal structures of tRNAs that bind to specific aaRSs are used here to prove the definition of “anticodon-amino acid” “relatedness”.
Miller [17] experiments with the abiotic synthesis of amino acids under conditions similar to those of the primitive Earth [17,18] and the amino acid composition of carbonaceous meteorites [19] support [GADV]-protein world hypothesis, which is based on pseudoreplication of [GADV]-proteins [1]. Generally, GAVD amino acids are accepted as the first amino acids to be created and they occupy the most thermodynamically stable complementary codons [20]. The second base from these codons is taken here for first nucleotideamino acid paring (g-G, c-A, a-D and u-V), which resulted in the “monobase codon-amino acid” phase of the genetic code evolution. The intermediate “relatedness” was designed based on above mentionned idea that the RNA bond formation was driven by peptide bond between two amino acids with each interaction being specific for a nucleotide. For example, I found in the human proteome 985 hits for GGGGGGGGGG, 658 hits for GVGVGVGVGV, 496 hits for GAGAGAGAGA and 285 hits for GDGDGDGDGD. It means that glycin preferentially reacted with other glycin in prebiotic GAVD soup; GG peptide bond was the most accessible for guanine nucleotides, so gg intermediate code was reserved for G. The human proteome served as a sample of the proteome, giving order of the peptide sequence hits. The hits depict how amino acids reacted at prebiotic pool. Interestingly, it is in accordance with the amino acid hydrophaty and modern codon table. Finally, the final phase was illustrated analogically, for example, the highest number among GXGXGXGXGX (X=all 20AA) peptides was obtained for GGGGGGGGGG, so glycine connected to di-guanine nucleic acid preferentially reacted with other glycine connected to mono or diguanine nucleotide (gg-G + g-G = ggg-GG) - “tribase codon-amino acid” phase of creation. These three phases were named according to Crick [21] who postulated that the evolution of the genetic code involved three phases:
I. The primitive code
II. The intermediate code
III. The final code, as we have it today [21].
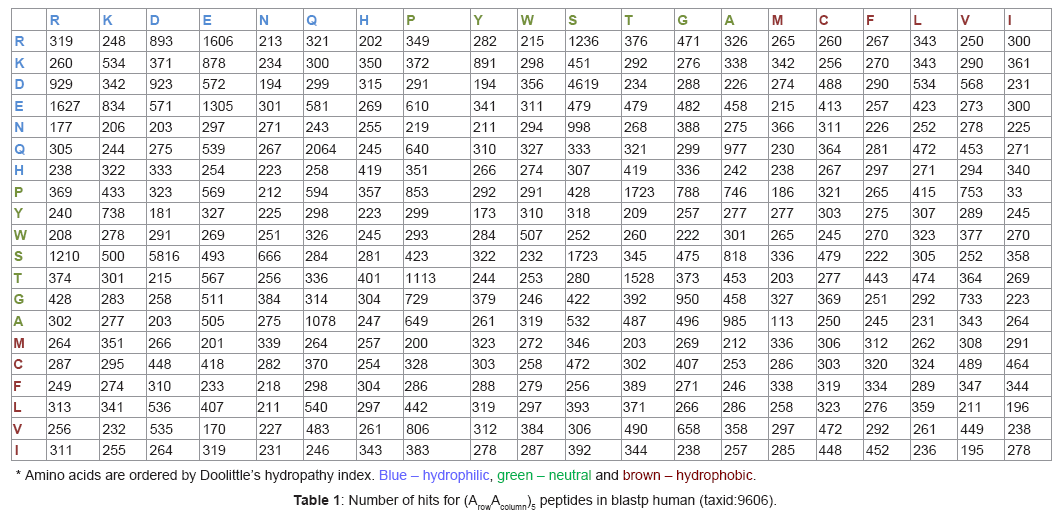
The Basic Local Alignment Search Tool (BLAST), the program which compares nucleotide or protein sequences to sequence databases and calculates the statistical significance of matches, was used to infer evolutionary relationships between RNA and peptide sequences (http://blast.ncbi.nlm.nih.gov/). The peptide sequence has been entered into “blast query sequence” and “non-redundant protein sequences (nr)” and “human (taxid:9606)” has been selected at “choose search set” and program was started using blastp algorithm. Searching was done for decapeptides which included five same peptide bonds for each amino acid including itself, with exception of selenocysteine (U) and pyrrolysine (O), (Table 1). For example, tendency to create stable peptide bond between glycine and alanine was explored by number of matches for “GAGAGAGAGA” peptide and for “AGAGAGAGAG” peptide (496 and 458 hits, Table 1).
Aminoacyl-tRNA synthetases complexed with cognate tRNAs were visualized using RCSB Protein Data Bank, view in 3D (Jmol), (http://www.rcsb.org/)
Analysis of the genetic code
Glycine, alanine, aspartic acid and valine are accepted as the first amino acids to be created. The order of the nucleotide triphosphates according to their solubility is as follows: ATP, CTP, UTP, and GTP. The most stable RNA triplets are ggc and gcc (28.3 Kcal/mole), which are followed by ggg and ccc (26.8 Kcal/mole) [20]. These characteristics are the basis of the primitive “monobase codon-amino acid” code, where the most abundant amino acid residues (AA), glycine and alanine, are related to g and c. Alanine is first hydrophobic amino acid residue in Kyte-Doolittle Hydrophaty index, so it was evolutionary convenience to be paired with better soluble pyrimidine base to produce “c-A” and the neutral glycine residue was paired with the least soluble purine base to produce “g-G”. According to above mentioned theory, the most abundant amino acid residues, glycine and alanine, created the first peptide bonds at prebiotic pool, which catalyzed RNA bonds formation. The AA abundance and RNA stability are reasons for the evolution of “c-A” and “g-G” “relatedness”. According to modern codon table, the most hydrophilic residue, aspartic acid, is related to most soluble adenine “a-D”, and the most hydrophobic amino acid residue, valine, is most closely related to uracil “u-V”. Looking over modern codon table, alanine is related to neutral AA and histidine to hydrophilic AA, so all AA in Kyte-Doolittle Hydrophaty index are shifted to left, and for better understanding, the “g-G”, “c-A”, “u-V”, “a-D” relatedness are colored, blue – hydrophilic, green – neutral and brown – hydrophobic (Table 1). VG and GV are the most conserved of the peptide bonds between V and the other amino acids from the group of G, A, V, and D, so the “dinucleotide” relatedness has been created according to the following: g-G + u-V = gu-VG (chemical equation summary) or gu-V (the relatedness writing), and it is in accordance with modern codons (Figure 1). Based on the valine “relatedness” to “u”, valine has been replaced by the more hydrophobic isoleucine in the case of the “au” dinucleotide (au-I), and by the less hydrophobic leucine in the case of uu-L and cu-L(Figure 1). Glycine, which is likely the most abundant amino acid in the prebiotic soup, preferentially forms peptide bonds with itself in among G, A, V, and D, according to the following: 2 x g-G = gg-GG or gg-G, and it is in accordance with modern codons (Figure 1). Alanine and aspartic acid formed the most evolutionary conserved peptide bonds with itself too, but cc-A and aa-D “relatedness” were adopted later by proline (cc-P) and lysine (aa-K) (Figure 1). Lysine is the second most hydrophilic neighbor of aspartic acid in Doolittle’s hydropathy index; the substitution is expected despite the opposite charge of K. In contrast, proline is quite far from alanine in Doolittle’s hydropathy index, which indicates that the amino acid group “Y, W, S, T”, placed between P and A in Doolittle’s hydropathy index, occurred later. Generally, P is accepted as the fifth appeared amino acid. So one can hypothesize about [GAVDP] protein world. The other most conserved amino acid peptide bonds created in the G, A, V, and D prebiotic pool for alanine and aspartic acid are GA and GD, so that g-G + c-A = gc-AG and g-G + a-D = ga-DG or gc-A and ga-D formed as we recognize them today in tribase codons. In the case of aspartic acid, more hits are observed for VD bonds and “relatedness” u-V + a-D = ua-DV, so probably ua-O/D (O = Pyrrolysine) was present at “dibase codon-amino acid” code, until it was reserved for tyrosine and stop codons. Aspartic acid, glutamic acid and glutamine have the same hydropathy index (HI = -3.5); there is a high probability that the intermediate “dibase” code did not distinguish between the negatively charged amino acids and shared one codon for aspartic and glutamic acid, ga-E/D, and polar uncharged glutamine assumed a different codon, ca-Q/D. Interestingly, serine and threonine replaced both alanine and glycine in the “dibase-codon” world and uc-S/A, ac-T/A, ag-S/G as we recognize today. Serine is a little more hydrophilic than threonine, so it adopts uc-S and threonine ac-T. Arginine is placed at the hydrophilic end, far from glycine, so it seems that arginine displaced serine or threonine later when the other forces were more important than the hydropathic similarity. Our first analytical step shows that the “dinucleotide-amino acid relatedness” regime naturally occurred primarily under prebiotic conditions, and hydropathic similarity played the main role at this stage of the evolution of the genetic code. The synthesis of prebiotic RNA bonds was influenced by the creation of prebiotic peptides, and everything originated from the “primitive relatedness” g-G, c-A, a-D and u-V. A number of hits of conserved fossil peptide sequences indicated the tendency of the peptide bonds created in the G, A, D, and V pool and when we accept “relatedness” between the creation of peptide and the nucleotide bonds, it is possible to depict the structure of the RNA c and their concentration in the prebiotic pool. One can hypothesize that prebiotic peptides and oligonucleotides participated on the same reaction [13-15], and despite the fact that evolutionary latter they split, proteins were specialized for catalysis and nucleic acids for coding, the relatedness is still conserved. The “dinucleotide-A” relatedness and the prebiotic peptide concentration (number of hits) estimate which and how many amino acids were involved in the reactions. According to quantity of prebiotic peptides and the “dinucleotide-amino acid relatedness”, it is possible to order the first fifteen amino acids into the chronology of their appearance as the first coded amino acids. For example the most concentrated cc-A was substituted by cc-P relatedness, that prove first proline appearance from next neutral AA (P,Y,W,S,T). The resulting order is as follows: “G,A,D,V,P,K,U,O,I,S,L,E,Q,T,R” (see Figure 1). Arginine does not follow hydropathy similarity, it was placed to the end. The final “tribase” codon evolved from the “dibase” codon and, again, peptide bond formation has been involved in its creation. For example, the creation of RD peptide bonds influenced RNA oligonucleotide formation as follows: cg-R + a-D = cga-RD, cg-R + ga-D = cgga-RD, ag-R + a-D = aga-RD, ag-R + ga-D = agga-RD. The process is illustrated in Figure 2, where it is possible to see that aspartic acid, serine, glutamine, threonine, proline, arginine, and glutamic acid have been preferentially involved. It is possible to estimate from the reactivity between amino acids that “gau” is the first tribase codon for aspartic acid, “uca, ucg” are the first tribase codons for serine, “acc” is the first tribase codon for threonine, “cgg, agg” are the first tribase codons for arginine and “gaa, gag” are the tribase codons for glutamic acid. With careful attention, one basic difference between Figures 1 and 2 can be detected. C-terminal amino acids are related to the intermediate code formation; this is illustrated by the GV peptide bond in which g-G + u-V = gu-VG or gu-V (Figure 1), which is contrary to the final code where the N-terminal amino acid is involved, and by the DS peptide bond, in which ga-D + uc-S = gauc-DS. This fact simply shows that it was created in the latter stage of the evolution by different mechanism and the triplet code has two letters related to the N-terminal amino acid and only one letter for the C-terminal amino acid, therefore gau is related to D and not to S or V.

Figure 1: “Mono-dibase codon-amino acid” logic. Order of hits (from the Table 1) for prebiotic peptides (AAAAAAAAAA, A=G,A,V,D) in blastp human (taxid:9606). Blue – hydrophilic, green – neutral and brown – hydrophobic.

Figure 2: “Tribase codon-amino acid” logic. Order of hits (from the Table 1) for prebiotic peptides (AAAAAAAAAA, A=amino acid) in blastp human (taxid:9606). Blue – hydrophilic, green – neutral and brown – hydrophobic; red – “tribase codon-amino acid”.
Our third analytical step will include a study of the anticodon reading by aminoacyl-tRNA synthetases. Figure 3 shows a solved crystal structure of glutamyl-tRNA synthetase complexed with tRNA(E). The glutamyl-tRNA synthetase is a class-I enzyme that can catalyze the initial amino acid activation reaction only in the presence of the cognate tRNA [22]. The loop of complexed tRNA(E) contains c534u535c536 anticodon nucleotides, and the anticodon-binding domains of EaRS contains “A431QPLRAALTG SL442ETPGL447FEI LALLGKERAL RRLERALA468“ sequence. Application of the “dibase codon-glutamic acid” logic explains how the synthetase recognizes the anticodon (“cuc”,”uuc”): SL442 recognizes uc cu/uu, and L447F recognizes cu/uu uu; serine provides the read-through for the second and third anticodon letters (uc), and leucine, together with phenylalanine, provides the read-through for the first and second anticodone letters (cu/uu-L, uu-F). SL442 and L447F reading sequences are followed by E, which is a charged amino acid that has opposite “relatedness” ga-E. Figure 3 clearly shows that the c534u535c536 anticodon bases are inserted into a level between the SL442 loop and the L447F α-helix. Figure 4 shows the crystal structure of cysteinyl-tRNA synthetase bound to tRNA(C). Two currently known codons for cysteine are “ugc” and “ugu”, so the first two letters of the anticodon are “gc” and “ac” and “dibase codon-amino acid relatedness” translation will lead us to look for A and T. As we know from above results, the triplet code has been evolved from the duplet code, so searching for the last two letters of the anticodon “ca-Q” will be more important. The anticodon-binding domain of CaRS contains “Q421QRLDARKAK DWAAADAARD RLNEMGIVLE DGPQGTTWRRK461“ sequence. The crystal structure of the complex revealed that the anticodon “g34c35a36” is identified by the Q454GTT sequence. Using the “relatedness” translation, g-G and ca-Q are responsible for the “gca” anticodon read-through and ac-T with ca-Q are responsible for the “aca” anticodon read-through. A sequence alignment of the most diverse CaRS group members (see Figure 5) shows that the translation process will be more variable. The “relatedness” g-G - ac-T is much more conserved then g-G - ca-Q or ac-T - ca-Q, so the GT dipeptide can read “gca”, and TT/TQ can read “aca”. Interestingly, the substitution of TT/TQ by TS/SK is still functional, which indicates the importance of the second letter in the “dibase codon-amino acid relatedness”, and confirms a preexistence of “monobase codon-amino acid relatedness” (see Figure 5). The GSK amino acid sequence translated based on “monobase codon-amino acid relatedness” means g-G, (t)c-S, (a)a-K or so “gca” anticodon. Another interesting point is that tryptophan is conserved next to the “reading sequence” and it has the same “double letter codon ug” as cysteine, which is the charged amino acid to Ca-RS. When we return to the expected results from the “dibase codon-amino acid relatedness” translation, the expected alanine residue was not identified in the short “reading” sequence, but the CaRS-tRNA complex structure shows that alanine is used to pull down the U-turn of the tRNA anticodon into the cavity of the CaRS C-terminal α/β domain (Figure 4). The “dibase codon-amino acid” translation for A, R and Q is “gc”, “cg” and “ca”, the “monobase codon-amino acid” translation for A, R, K and D is “c”, “g”, “a” and “a”, and the cavity is full of these amino acids: “Q421QRLDARKAKDWAAADAARD”. It seems that the indirect orientation of “g34c35” is controlled mainly by alanine and “c35a36” mainly by glutamine (Figure 4).

Figure 3: T. thermophilus EaRS•tRNA(E) complex. L442 and opposite L447 recognize “c534u535”. RCSB Protein Data Bank, 1N78.

Figure 4: Escherichia coli CaRS•tRNA(C) complex. Using the “relatedness” translation, ac-T and ca-Q are responsible for the “c35a36” read-through and gc-A and cg-R are responsible for the “g34c35” read-through. RCSB Protein Data Bank, 1U0B.

Figure 5: Part sequence of anticodon-binding domain of cysteinyl tRNA synthetases (CaRSs). Sequence Alignment of the most diverse group members.
Prediction of anticodon reading sequences inside the aminoacyl-tRNA synthetases by “codon-amino acid” relatedness
As described above, the one “di-base relatedness” can be substituted with two “mono-base relatedness” what complicates the potential modelling of the protein ↔ nucleic acid interaction process, but it can still be helpful for the identification of the protein sequences for nucleic acid “reading”. The Thermotoga maritima tryptophanyl-tRNA synthetase (WaRS) has only one codon, and the enzyme is relatively small (328AA). The C-terminal domain follows the sequence:
“E205ISEKELEQTILRMMTDP222ARVRRSDP 230 GNP233ENCP237VW239KYH242QAFDISEEESKWV-WEGCTTASIGC-VDCKKLLLKNMKRKLAP 283 IWENFRKIDEDP295HYVDDVIM-EGTKKAREVAA-KTMEEVRRAMNLMF 328”. The “dibase codonamino acid relatedness” translation of the “cca” anticodon will lead us to look for ca-Q, ca-H, ac-T and cc-P, or the “monobase codon-amino acid relatedness” translation of “cca” provides c-A, c-S, c-T and aa-K, aa-N, a-D, a-E letters. This is quite a lot of combinations; however, it helps that the “reading sequence” is usually close to the charging amino acid because it has opposite relatedness as its anticodon, which is tryptophan or the cysteine with the same “double-codon” can be considered. The potential amino acids are coloured in the above C-terminal domain sequence and W239 with P233, P237 on one side and H242QA sequence on other side appear to be potential “cca” reading amino acids. The whole C-terminal domain has only two glutamines and two histidines, so H242QA and P237 appear to be the most important. Determining the sequence alignment (see Figure 6) indicates that P233 and H242 are the most important anticodon-reading amino acids. This can be considered to be a result of “relatedness” analysis of the primary sequence. The crystal structure of WaRS is accessible (Figure 7), so we can illustrate anticodon reading, which can be accomplished without complex crystallization with cognate tRNA. The “cca” anticodon nucleotides are navigated by oppositely oriented proline residues, cc-P233 and cc-P237, and by aa-N235 placed up the prolines, into the anticodon domain cavity, where it is oriented by ca-H242 on one side and by (t) c-S254 plus aa-K253 on the opposite side. The opposite oriented tryptophan residues, W239 and W256 are also internally exposed into the cavity to provide smooth movement within; they have the exact contrary “relatedness”, ugg-W (see Figure 7). C236, C259, C266, C239 cysteine residues with contrary “dinucleotide-AA relatedness” do not interrupt the anticodon reading but hold four iron atoms inside the cavity by their S atoms. Perhaps it only stabilises the cavity structure, because they showed WaRS is hyperthermophilic, or the irons fix the anticodon position [23].

Figure 6: Part sequence of anticodon-binding domain of tryptophanyl-tRNA synthetases (WatRS).

Figure 7: Thermotoga maritima tryptophanyl-tRNA synthetase. Using the “relatedness” translation of “cca” anticodon, cc-P233 and cc-P237 are responsible for the “cc” read-through; ca-H242 and (t)c-S254 plus aa-K253 are responsible for the “ca” read-through; aa-N235 completes the prolines to “cca” read-through; and the codon relatedness ugg-W does not interrupt the read-through. RCSB Protein Data Bank, 2G36.
Various possible scenarios, which lead to the emergence of the genetic code, have been indicated in the introduction. However the presented results imply that the strongly supported RNA world theory is not clear. The results support researchers that suggest that a full-blown RNA world in fact never existed [24]. Instead, very early coevolution of nucleotides and amino acids evolved to produce a “ribonucleopeptide” world where ribonucleotides were larger, because the RNA nucleotides were more stable than peptides (for example Prokaryotic ribosomes are composed of 65% ribosomal RNA and 35% ribosomal proteins), but all rules have been already designed based on the GAVD time period when interactions of emerged amino acids with nucleotides leaded to short RNA synthesis. The bioinformatic evidence presented here, that the first nucleotide bonds were connected to the peptide bonds and that distribution of prebiotic peptides still correlates with DNA code, confirms the “relatedness” picture that has been designed by Woese [16] and other previous theories [24]. Lehmann and coworkers [11] simulated the polymerization of amino acids along RNA templates and concluded that a system of four codons (gnc, n= g, c, u, a) and four amino acids (G, A, V, D) could be a plausible original genetic code. Independently, Higgs [25] proposed four column triplet code (gnn, n= g, c, u, a) and four amino acids (G, A, V, D) as the earliest genetic code. The presented work changes the scenario, four amino acids (G, A, V, D) “related” to four nucleotides (g, c, u, a) created the first peptides and the first RNA oligomers were synthesized along the peptide templates, [GADV]-protein world [1] or better [g-G,c-A,u- V,a-D]-ribonucleopeptide world. It is probable that ribonucleopetides catalyzed first metabolic reactions and latter they split, proteins were specialized for catalysis (20 AA - better variability) and nucleic acids for coding (DNA - better stability).
As have already Woese, [26] pointed out, similar polar properties are required for amino acids coded by the same base in the second codon position, hydropathic similarity played the main role at the evolution of the intermediate (dibase) code (Figure 1, Table 1). The third letter adition to triplet code creation, in difference, is independent on hydropathic similarity, but still follow the peptide bonds formation in the enviroment. Lehmann and Libchaber, [27] analyzed the anticodoncodon association within the ribosome decoding center. Interestingly, they pointed out stability of the base pair at the second position of the anticodon and fact that the canonical U-turn of the tRNA anticodon loop contributes to the degeneracy at the third position of the codon, what is in accordance with the presented theory of the genetic code evolution.
Despite many structural, kinetic and thermodynamic studies that have been published [28,29], all the forces that relate nucleic and amino acids are unknown, so it is still not sufficient to consider all of the components together, such as energy, frequency and propensities of amino acid-nucleotide interactions, water-mediated interaction, or other physicochemical and stereochemical forces. Based on Woese’s work [16], when we do not obtain information from “all the interactions of which a molecule is capable”, we cannot predict or calculate universal recognition rules. A complete equation is not achievable today, but it is possible search a correlation by bioinformatical tools and efficiently evaluate empirical data from structures in PDB [30] or from amino acid-nucleotide affinity chromatography [12], chip-seq data [31], or data from other sources.
This contribution is the result of the project implementation: Centre of excellence for white-green biotechnology, ITMS 26220120054, supported by the Research & Development Operational Programme funded by the ERDF.