Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2013) Volume 6, Issue 3
Molecular pathways regulating melanoma initiation and progression are potential targets of therapeutic development for this aggressive cancer. Identification and molecular analysis of these pathways in patients has been primarily restricted to targeted studies on individual proteins. Here, we report the most comprehensive analysis of formalin-fixed paraffin-embedded human melanoma tissues using quantitative proteomics. From 61 patient samples, we identified 171 proteins varying in abundance among benign nevi, primary melanoma, and metastatic melanoma. Seventy-three percent of these proteins were validated by immunohistochemistry staining of malignant melanoma tissues from the Human Protein Atlas database. Our results reveal that molecular pathways involved with tumor cell proliferation, motility, and apoptosis are mis-regulated in melanoma. These data provide the most comprehensive proteome resource on patient melanoma and reveal insight into the molecular mechanisms driving melanoma progression.
Keywords: FFPE tissue; Melanoma; Proteomics; Quantitative; Mass spectrometry
In current clinical practice, human tissue specimens are routinely fixed in formalin before histological analysis and archiving. Due to the stability of these tissues following fixation, many health care organizations have acquired vast archives of Formalin-Fixed, Paraffin- Embedded (FFPE) tissues. These tissues are typically stored with information associated with patient diagnosis and outcome, thus they have been viewed as ideal specimens for the identification of biomarkers that may aid in disease diagnosis, prognostication and personalizing treatment regimens. In addition to their use for identifying biomarkers, archival tissues have also been useful in characterizing the biochemical and cellular processes involved in disease initiation and progression. During fixation, formaldehyde reacts with N-terminal, arginine, cysteine, histidine, glycine and lysine amino acids to form methylol groups and methylene bridges [1]. Methylene bridges, or covalent cross-links, inactivate or immobilize proteins depending on where the linkage occurs. Chemical fixation of proteins in tissues presents a significant challenge for protein or nucleic acid extraction for research. One such area of research presented with this challenge is the high throughput analysis of protein levels for the presence of biomarkers in disease development. These proteomic approaches for measuring protein levels in FFPE tissues have largely relied on label-free mass spectrometry as techniques involving chemical labeling are hampered by inefficient formaldehyde cross-linking reversal at amino acids reactive with chemical labels for proteomic studies.
In this study, we report the most comprehensive to date label-free, quantitative mass spectrometric analysis of protein levels in benign nevi, primary melanoma and metastatic melanoma from FFPE patient tissue samples. Cutaneous malignant melanoma is an aggressive neoplasm arising from melanocytes in the skin. While malignant melanoma is not the most common form of skin cancer, it is the most deadly and incidences have been steadily increasing worldwide [2]. According to the American Cancer Society, an estimated 76,250 new cases of melanoma will be diagnosed in 2012 and approximately 9,180 people will die because of this disease [3]. This mortality rate is at least partially due to the apparent chemoresistance of advanced stage melanoma cells [4]. Studies have shown that melanocytes are inherently resistant to apoptosis and that melanoma cells acquire additional antiapoptotic and pro-survival features during melanoma progression [5]. The application of cutting-edge profiling technologies may provide new insights into the molecular basis of chemoresistance in melanoma. Several profiling approaches have been used to characterize molecular features of melanoma cells and tissues, including gene expression profiling, proteome analysis and serum proteome profiling [6]. The majority of Mass Spectrometric (MS)-based proteomic studies have been limited to melanoma cell lines, cultured melanoma cells and patient serum samples. Recently, label-free MS proteomic studies of FFPE patient melanoma samples have been reported [7-9]. The ability to analyze actual patient samples provides the most relevant readout of the functional melanoma proteome. In one study, utilizing 24 patient samples (8 primary and 16 metastatic melanomas), 555 proteins were identified with a high confidence and a False Discovery Rate (FDR) of < 5% and 61 were found to be significantly differentially expressed between primary and metastatic melanoma tissues [7]. In other studies only one metastatic melanoma [8] or two (one nevus and one malignant melanoma) [9] patient samples were analyzed resulting in the identification of 935 proteins by Rezaul et al. [8] and of 888 proteins by Byrum et al. [9] with a FDR of <1% for both studies. Rezaul et al. [8] validated expression of four identified proteins using Immunohistochemistry (IHC) and Byrum et al. validated two [8,9].
In the current study, a total of 61 patient samples (25 nevi, 12 primary and 24 metastatic melanomas) were used for quantitative proteomic analysis, making it one of the most comprehensive FFPE proteomic studies for melanoma. We identified 1528 proteins with a high confidence (FDR <1%) among these samples, of which 171 significantly varied in abundance between benign nevi, primary melanoma and metastatic melanoma FFPE tissues. Protein levels were correlated to reported findings for malignant melanoma in the Human Protein Atlas (HPA) database, a repository of Immunohistochemistry (IHC) data for a multitude of tissues and cells assembled on tissue microarrays [10]. We found that 73% of the proteins identified by our quantitative proteomic analysis correlated to the HPA. Additionally, 41 proteins not analyzed in the HPA were significantly differentiated between benign and primary and metastatic melanoma. Of these, 33 have never been correlated with melanoma and thus may represent a panel of new putative markers for melanoma. The proteins identified in this study shed light onto the mechanisms of melanoma progression, which include melanoma cell proliferation, cell motility and resistance to apoptosis.
Tissue processing
Archival FFPE tissues were obtained from the University of Arkansas for Medical Sciences Department of Pathology. All studies reported here using de-identified, archived FFPE human patient tissue samples were approved by the University of Arkansas for Medical Sciences Institutional Review Board with approval #113003. In accordance to IRB approval #113003, all FFPE human patient tissue samples were sufficiently de-identified and determined exempt from consent as these studies used existing archived pathological specimens and not human subjects directly. Human tissue specimens were identified by two independent, board-certified pathologists. The following FFPE samples from skin biopsies were used for proteomic studies: 25 benign, 12 primary melanomas, and 24 metastatic melanomas. Cases were selected from original H&E stains. Specimens were cut into 10 μm sections on glass slides and then incubated at 58°C for 60 minutes. Sections were deparaffinized in xylene twice for 5 minutes then rehydrated in graded ethanol solutions (100% ethanol twice for 5 minutes, 85% ethanol twice for 1 minute, and 70% ethanol twice for 1 minute) and washed twice in purified water for 1 minute then air-dried. Tissue was collected with a needle to ensure that >95% of cells collected were cells of interest (i.e., melanoyctes or melanoma cells). To extract proteins, tissue was solubilized in 20 mM Tris, pH 7 / 2% SDS, and incubated at 90°C for 1 hour. Tissues were sonicated for 5 minutes using a Bioruptor UCD 200 (Diagenode) on high power with a 30 second on/off cycle. Formalin cross-linking was reversed at 65°C for 4 hours. Two micrograms of protein for each sample was resolved by 4-20% SDS-PAGE (Invitrogen pre-cast gels) and Coomassie-stained. Gel lanes were cut into 24 sections and subjected to in-gel trypsin digestion as described [9]. Protein-containing gel slices were destained in 50% methanol, 100 mM ammonium bicarbonate, followed by reduction in 10 mM Tris[2-carboxyethyl] phosphine and alkylation in 50 mM iodoacetamide. Gel slices were then dehydrated in acetonitrile, followed by addition of 100 ng porcine trypsin (Promega) in 100 mM ammonium bicarbonate and incubation at 37°C for ~14 hours. Peptide products were then acidified in 0.1% formic acid.
Mass spectrometry and protein identification
The approach used for mass spectrometric analysis of protein samples is largely as reported in Byrum et al. [9], which showed a high level of technical reproducibility (Pearson correlation >0.98 and p<0.0001). Tryptic peptides were analyzed by nanoflow LC-MS/MS with a Thermo Orbitrap Velos mass spectrometer equipped with a Waters nanoACQUITY LC system [11,12]. Tryptic peptides were separated by reverse phase Jupiter Proteo resin (Phenomenex) on a 100x0.1 mm column using a nanoAcquity UPLC system (Waters). Peptides were eluted using a 30 min gradient from 98:2 to 40:60 buffer A:B ratio. [Buffer A=0.1% formic acid, 0.05% acetonitrile; buffer B=0.1% formic acid, 75% acetonitrile.] Eluted peptides were ionized by electrospray (2.0 kV) followed by MS/MS analysis using collision induced dissociation on a Thermo LTQ Orbitrap Velos mass spectrometer. MS data were acquired using the FTMS analyzer in profile mode at a resolution of 60,000 over a range of 375 to 1500 m/z. MS/MS data were acquired for the top 15 peaks from each MS scan using the ion trap analyzer in centroid mode and normal mass range with normalized collision energy of 35.0. A total of 1528 proteins were identified by a Mascot (version 2.2.03) database search with the following parameters: precursor ion tolerance 5 ppm, fragment ion tolerance 0.65 Da, fixed modification of carbamidomethyl on cysteine, variable modification of oxidation on methionine, and 2 missed cleavages possible with trypsin. We searched the human ‘UniProtKB/Swiss-Prot’ database (http://www.uniprot.org/downloads) and we additionally used reversed sequences for higher confidence identifications. The Mascot results were uploaded into Scaffold 3 (version 3.00.01) for viewing the proteins and peptide information. A false discovery rate of 1% was used as the cut off value and spectral counts were exported into an Excel spreadsheet (Supplemental Table S1).
Quantitative analysis of protein levels
In order to determine significantly differentiating levels of proteins between benign nevi, primary melanoma, and metastatic melanoma, a label-free approach based on spectral counting was used [9,13-15]. A spectral count is the number of tandem mass spectra assigned to a given protein in a single gel lane [9,13].
Prior to statistical analysis, the spectral count data were first normalized in order to compare between samples, account for heteroscedasticity (log transformation), and to account for the relative amount of proteins in a given gel lane by calculating a Normalized Spectral Abundance Factor (NSAF) [13,16].
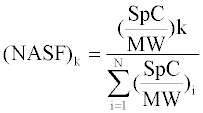
The NSAF for a protein k is the number of Spectral Counts (SpC) identifying a protein, k, divided by the protein’s Molecular Weight (MW), divided by the sum of SpC/MW for all N proteins in the gel lane [9]. Different proteins were identified among the 61 FFPE tissue samples and therefore, some proteins will contain a spectral count of zero for a particular sample. In order to allow for log transformation, Zybailov et al. [13] previously described a method for dealing with spectral counts of zero in the data set by replacing the zero values with a fractional value prior to the NSAF calculation. However, in the present study, instead of assigning a fractional value and treating the zero values differently from the rest of the data values, we opted to shift the entire data set by adding 0.1 to every value. In this way every value is treated equally prior to the calculation of ln(NSAF). As is commonly done in microarray data sets, the data was scaled to ensure the medians of all distributions were equal and centered to ensure the standard deviations of all distributions were equal, allowing for more robust statistical testing [17].
The FFPE tissues were analyzed in three sample groups; benign, primary melanoma, and metastatic melanoma. The non-parametric Kruskal-Wallis test was employed to determine significantly differentiating proteins among the three sample groups (Supplemental Table S2). Kruskal-Wallis indicates when a protein is significantly different, but does not differentiate where the significance lies between groups. Therefore, multiple Mann-Whitney U tests were calculated for each Kruskal-Wallis significant protein to determine group significance. Mann-Whitney with Bonferroni correction was used to test the null hypothesis of no difference between two groups; benign and primary, benign and metastatic, and primary and metastatic (Supplemental Table S3). Fold Changes (FC) were also calculated using the average normalized spectral counts for each group. A recent review by Wu et al. [16] demonstrated the NSAF normalization method using spectral counts has greater precision over MS/MS ion intensity based calculations; however, fold changes with spectral count data are seriously underestimated and should not be the sole indicator of protein expression. Therefore, fold changes were used only as a filtration method. Proteins with a spectral count ≥ 5, a Mann-Whitney p-value < 0.05, and a fold change greater than 2 were considered to have the most significance (Supplemental Table S4). Proteins differing in levels are illustrated in volcano plots and listed in Supplemental Table S5.
Pathway-Express from Onto-Tools (http://vortex.cs.wayne.edu/projects.htm [18]) was used to identify known pathways containing the proteins of interest. The significant protein list was uploaded into Pathway Express along with a reference list containing all of the 1528 identified proteins from the proteomic data set. The default parameters were used for the analysis including a hypergeometric distribution, p-value threshold of 5%, impact factor threshold of 50%, and all regulatory efficiencies of 1.0 with the exception of inhibition and repression of -1.0.
To further validate our findings, the Human Protein Atlas (HPA) database was searched with the significantly differentiating proteins to identify their expression in human malignant melanoma tissue. The HPA project generates protein expression profiles based on Immunohistochemistry (IHC) with 1 to 4 antibodies for each protein [10]. They report the IHC staining as strong, moderate, weak, or negative as percentages. We added the percentages for strong, moderate, and weak and only report positive or negative staining in Supplemental Table S6 for proteins determined to be significant from our proteomics study. Since the HPA contained IHC results for malignant melanoma but not for nevi, we were unable to directly correlate up- or down-regulation of proteins relative to benign tissues. Up-regulation of proteins in primary and metastatic melanoma in the proteomic data set was correlated to positive staining in the HPA database and the down-regulation of proteins was correlated to negative staining.
The present study is a comprehensive proteomic analysis using patient FFPE tissues to study relative protein levels in different stages of melanoma. Proteins were isolated and relative levels were determined by label-free mass spectrometry between benign nevi, primary melanoma, and metastatic melanoma archival samples (figure 1). High resolution mass spectrometry identified 1528 proteins with a high confidence (FDR < 1%) from 61 patient samples (Supplemental Table S1 and Supplemental Figure S1), where abundances of 171 proteins were found to be significantly different between the three sample groups by Mann Whitney (p-value < 0.05), fold change > 2, and spectral count ≥ 5 (Supplemental Table S4). Relative to the proteomic study by Huang et al. [7] that identified 61 proteins significantly differentially expressed between primary melanoma and metastatic melanoma, we identified 5 (HNRPL, FTL, COX4I1, DCN, LUM) of the 61 proteins as differentially expressed and also observed 166 additional proteins as differentially expressed. Our study is the largest proteomic data set reported thus far for FFPE melanoma and offers many insights into the progression of the disease from benign nevi to primary melanomas and finally to the most aggressive form, metastatic melanoma.

Figure 1: Quantitative proteomic analysis of FFPE melanoma: Melanocytes from benign FFPE skin biopsies (25 total) and melanoma cells from primary (12 total) and metastatic melanoma (24 total) FFPE biopsies were isolated by needle dissection. Protein extracts were resolved by SDS-PAGE, visualized by Coomassie staining, excised as 24 bands per lane, and subjected to in-gel trypsin digestion. Tryptic peptides were analyzed by LC-MS/MS and relative protein levels were determined by spectral counting [9].
A hierarchical cluster of all 61 samples and their significant proteins clearly separated the benign nevi, primary melanoma, and metastatic melanoma tissues into three distinct clusters (Figure 2). Interestingly, a discrete cluster of proteins showed increased expression in primary melanoma compared to both benign nevi and metastatic melanoma, including RAB21, LAMP1, EIF4A3, HEXB, SERPINB1, SLC25A11, HNRNPF, EMD, HP1BP3, PTBP1, TPP1, and TUBB2A. These proteins may be potential targets for early stage tumorigenesis. A few particularly interesting proteins are Rab21, Lamp1, and eIF4A3. The small GTPase Rab21 has been shown to positively regulate integrin-mediated cell adhesion and motility; overexpression of Rab21 also stimulates cell migration and adhesion of cancer cells to collagen and bone [19]. Lamp1 is a lysosomal membrane glycoprotein that has been implicated in melanoma metastasis [20] and that may be a target for cancer immunotherapy [21]. eIF4A3 is an ATP-dependent RNA helicase, a component of the exon junction complex involved in mRNA expression [22] and may be a serum biomarker for pancreatic cancer [23].

Figure 2: Hierarchical cluster of significantly differentiating proteins: An unsupervised cluster of both FFPE biopsies and significant proteins shows clear separation among Benign (BS), Primary Melanoma (PM), and metastatic Melanoma (M) patients. A sub-cluster of proteins shows increased expression in the primary samples compared with both benign and metastatic (in brackets). Red data points indicate increased protein expression, while green indicates decreased expression.
Proteins that were significantly differentiated between each group are illustrated in volcano plots (Figure 3). There were 113, 60, and 21 significant proteins with a Mann-Whitney p-value < 0.01 and a fold-change > 2 between benign nevi and primary melanoma, benign nevi and metastatic melanoma, and primary melanoma and metastatic melanoma samples, respectively (Supplemental Table S5). Many of these proteins were found to be involved in known pathways involved in cancer progression including MAPK signaling, focal adhesion, adherens junctions, regulation of actin cytoskeleton, Extracellular Matrix (ECM)-receptor interaction, and apoptosis (Figure 4). These pathways were identified by Pathway-Express from Onto-Tools, which links to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, a collection of databases integrating pathway, genomic, proteomic, and ligand data [18,24]. The pathways and their associated proteins from the proteomic data set are shown in figure 5. Several mis-regulated proteins are involved in multiple pathways and therefore, have a major impact on tumor progression. Further investigation of this data set revealed several other proteins not annotated by Pathway-Express including: Proline/Arginine-Rich End Leucine-Rich Repeat Protein (PRELP), lumican and decorin, ferritin heavy-chain (H-ferritin), membrane-organizing extension spike protein (moesin), thymosin beta-10, and Prothymosin-α (PTMA), which are involved in proliferation, cell-ECM interactions, cell motility and apoptosis.

Figure 3: Volcano plots of significantly differentiating proteins: The negative log (base 10) of the Mann Whitney U p-values are plotted on the y-axis and the log (base 2) of the fold change are plotted on the x-axis. A) Metastatic melanoma versus benign. B) Primary melanoma versus benign. C) Metastatic melanoma versus primary melanoma. The red data points indicate proteins with a p-value < 0.01 and a fold change > 2.

Figure 4: Pathways identified for significantly differentiating proteins: The number of significantly differentiating proteins associated with each pathway is displayed in the bar graph. The pathways with the most proteins have the highest mis-regulation.

Figure 5: Detailed pathways identified for significantly differentiating proteins: The major pathways that are mis-regulated in benign nevi, primary melanoma, and metastatic melanoma and their associated proteins are shown as a pathway map modified from the multiple KEGG pathways. The pathway map highlights proteins up-regulated in benign nevi in blue, primary melanoma in green, and metastatic melanoma in red.
We have been able to identify several proteins involved in pathways associated with survival signaling and cell motility in melanoma and have validated their expression by IHC data using the HPA database. The database contained IHC results for 130 of the 171 significantly differentiated proteins identified by our quantitative proteomic study (Supplemental Table S6). The patterns of protein levels observed in this data set were consistent with 73% (85/116) of the reported IHC data. A small group of 14 proteins had about 50% positive staining (ranging from weak to strong) and about 50% negative staining in the IHC data and so were not conclusive. Interestingly, we also identified 41 proteins not analyzed by HPA, 33 of which have not previously been correlated to melanoma progression. These proteins are currently pending analysis in the HPA database and include PRELP, Tmsb10, and serpinH1 (Supplemental Table S6).
Metastatic melanoma is an aggressive form of cancer given that tumor cells frequently disseminate to multiple organs, such as brain, lungs, liver, and/or bone, making surgical treatment ineffective [25,26]. Metastatic melanoma is also non-responsive to chemotherapy, which is likely a consequence of intrinsic survival pathways in normal melanocytes [5] and acquired survival queues in melanoma cells [5]. Most epithelial cells respond to DNA-damaging agents or radiation by inducing apoptosis or halting the cell cycle until these cells can repair damage to their DNA. However, melanocytes are the photoprotectors of the skin and in contrast, respond to DNA damage by secreting melanin which protects neighboring keratinocytes from further damage [5]. Normal melanocytes are long-lived postmitotic cells that do not produce mitogens and therefore, depend on the release of keratinocyte-derived growth factors to stimulate cell proliferation [27]. The Mitogen-Activated Protein Kinases/Extracellular Signal-Regulated Kinase (MAPK-ERK) pathway plays an important role in the progression of melanoma by inducing melanoma cell proliferation independent of these growth factors and enhances cell survival and resistance to apoptosis [28-30]. Induced by ras oncoproteins, the MAPK-ERK signaling pathway activates a number of growth-promoting genes, confers anchorage independence and loss of contact inhibition [31]. We found an inhibitor of MAPK activation, Gng12, to be expressed in benign nevi and significantly decreased in primary or metastatic melanoma. This small GTPase is a negative regulator of ras activity [32], and presumably inhibits the activation of the MAPK signaling cascade in nevi [33,34]. Decreased levels of Gng12 in primary and metastatic melanoma samples represent the loss of ras inhibition, possibly contributing to sustained ras signaling in these tissues. Gng12 was not analyzed by HPA and is unique to our dataset. Additionally, Mapk1 was found to be up-regulated in primary and metastatic melanoma samples relative to nevi, which is consistent with previous findings that the MAPK pathway is an important component of melanoma progression [35], immune evasion [36] and inhibition of apoptosis downstream of cytochrome c release [30] (Figure 5). Mapk1 was also validated by HPA with 89% positive staining for malignant melanoma.
Oncogenic ras mutations activate both MAPK-ERK and the phosphatidylinositol-4,5-bisphosphate 3-kinase/ serine/threonine protein kinase (PIK3-AKT) pathways. MAPK-ERK is also activated by mutations in the BRAF gene, while PI3K-AKT is activated by loss of the tumor suppressor gene PTEN. These mutations occur early in melanoma pathogenesis and are preserved throughout tumor progression [37]. The binding of receptor kinases at the plasma membrane activates PI3K, which converts PIP2 to PIP3. PIP3 then activates protein kinase B (PKB) - AKT with downstream effects on multiple targets involved in cell proliferation, migration, and survival [5]. Survival signals are induced by several receptors mediated mainly by the PIK3/AKT pathway. Therefore, up-regulation of this pathway may contribute to metastatic melanoma resistance to chemotherapy and drug treatments [38] (Figure 5). We identified upstream regulators of this pathway that were unique to our proteomic data set including Gng12, Gna12, and Rac3.
In addition to the growth-stimulatory effects of MAPK-ERK and PIK3-AKT activation, Mapk1 also plays a role in the melanogenesis pathway, which is responsible for the synthesis of melanin. Mapk1 induces Mitf, which promotes the transcription of Tyrosinase (TYR), Tyrosinase-Related Protein 1 (TYRP1), and Dopachrome Tautomerase (DCT) proteins in order to convert tyrosine to melanin, where melanin is transferred to keratinocytes (Figure 5). Melanin is a photoprotectant that absorbs harmful UV-radiation, transforms the energy into heat, and protects cells from indirect DNA damage that is responsible for metastatic melanoma [39]. However, melanin can also produce active radicals that can damage DNA. The protective aspect of melanin in dark skin is a result of the high concentration and densely packed organelles that shield the nucleus. Conversely in light skin, the production of radicals supports the development of metastatic melanoma [40]. MAPK-ERK, PIK3-AKT and melanogenesis promote cell survival, likely contributing to drug resistance in melanoma.
The inactivation of apoptosis is a hallmark of cancer [31] and also leads to enhanced melanoma cell survival and increased resistance to chemotherapeutic agents [5]. Melanoma cells also likely benefit from autocrine- and paracrine-induced survival signaling [41] resulting in cancer cells with extreme drug resistance. In the current proteomic data set we have identified three pro-survival and anti-apoptotic factors, L- and H-Ferritin and PTMA, at increased levels in primary and metastatic melanoma relative to benign nevi. H-Ferritin showed 100% positive staining in malignant melanoma by HPA; however, L-Ferritin only showed 36% positive staining. PTMA was not analyzed by HPA and is unique to our proteomic data set. PTMA has previously been shown to inhibit apoptosome formation and ferritin functions to increase resistance to oxidative stress. Ferritin is a globular protein which functions as the major iron storage component of mammalian cells [42]. Ferritins are composed of both L- and H-ferritin subunits which are assembled in different proportions, likely in a tissue-specific manner [43], to form various ferritin isoforms. Interestingly, ferritin (particularly L-ferritin) has been shown to contribute to melanoma cell growth and insensitivity to oxidative stress [44]. Up-regulation of H-ferritin by NF- κB has been shown to inhibit apoptosis by suppressing reactive oxygen species accumulation [45]. We have also identified increased levels of anti-apoptotic PTMA in primary and metastatic melanoma tissues. PTMA is a nuclear oncoprotein that inhibits procaspase-9 activation by preventing apoptosome formation [46]. Initially referred to as a “thymic peptide,” PTMA is a ubiquitously expressed regulator of cell cycle and immune response [47]. PTMA has been found to be overexpressed in human bladder cancer and its overexpression is associated with poor prognosis in breast, gastric, liver, prostate and head and neck cancers [47].
The regulation of actin polymerization and ECM-receptor interaction pathways may help prepare melanoma cells for tumor migration by disrupting cell-to-cell contact and by increasing cell motility [48]. Actin polymerization, a major component of cell motility, drives the extension of a protrusion and depends on the availability of actin monomers [49-51]. In this proteomic data set thymosin beta-4 (Tmsb4x) and thymosin beta-10 (Tmsb10), which function to sequester actin monomers for the process of actin polymerization [52], were found increased in primary and metastatic melanoma samples compared to nevi. Increased expression of Tmsb10 has been identified in melanoma cell lines and was correlated with metastatic potential [53,54]. After cell extension, new adhesions are formed to stabilize the protrusion, including the binding of collagen, laminin, and fibronectin to the Extracellular Matrix (ECM) [49]. Fibronectin has been implicated as a major player in metastatic melanoma and cell motility and was found up-regulated in the metastatic melanoma samples [55] (Figure 5). Additionally, the Ezrin-Radixin-Moesin (ERM) family protein moesin was found up-regulated in primary and metastatic tissues. Moesin functions as a cross-linker between the plasma membrane and actin cytoskeleton, regulating cell protrusions and movement [56]. It has been shown to play a role in melanoma cell polarization and contributes to invasion and metastasis [57]. We also detected a decrease in decorin and lumican in primary and metastatic samples, two abundant proteoglycan components of the skin ECM. Decorin has growth-suppressive activity which occurs via up-regulation of the CDK inhibitor p21 [58]. Lumican has been shown to inhibit melanoma progression in mouse models by regulating cell migration, proliferation and apoptosis [59]. Moesin, decorin, and lumican were all validated by HPA. Our findings suggest there is increased cytoskeletal remodeling and altered ECM in primary and metastatic melanoma tissues relative to benign nevi, which indicates that melanoma cells in these tissues, may be more mobile and have a greater potential for invasion and metastasis [60].
In conclusion, we present the most comprehensive to date proteomic study for melanoma progression using patient samples. Using an unbiased, high-throughput and quantitative label-free mass spectrometric approach for the identification of proteins from benign nevi, primary melanoma, and metastatic melanoma FFPE tissues, we identified a total of 1528 proteins. Of these proteins, 171 proteins were significantly differentiated between the three groups and the vast majorities (73%) of the proteins were validated by the HPA staining in malignant melanoma tissues. These proteins are involved in several pathways known to play major roles in cancer tumorigenesis. In primary and metastatic melanoma FFPE tissues, we identified decreased levels of proteins associated with cellular-ECM interactions and detected increased levels of proteins that promote cell motility, proliferation and survival. Our extensive study of archived human melanoma tissues presents a vast amount of putative biomarkers that may help clinicians with diagnosis, prognosis and treatment of metastatic melanoma.
The authors declare no conflict of interest with this work including relationships that could be construed as resulting in an actual, potential, or perceived conflict of interest with regard to this manuscript
The authors declare that they have no competing interests.
SDB, SKL, LM & SGM carried out the experiments and data analysis. SDB, SKL and NLA drafted the manuscript. WLC and AJT conceived of the study and participated in its design and coordination. AJT finalized the draft of the manuscript. All authors read and approved the final manuscript.
We would like to acknowledge the UAMS Proteomics Facility for mass spectrometric support and support from NIH grants R01DA025755, F32GM093614, UL1RR029884, KL2RR029883, P30GM103450, and P20GM103429. We would also like to acknowledge Yongmei Wu and Tatiana Leakey for technical assistance.