Journal of Physical Chemistry & Biophysics
Open Access
ISSN: 2161-0398
![]() +44 1478 350008
+44 1478 350008
ISSN: 2161-0398
![]() +44 1478 350008
+44 1478 350008
Research Article - (2017) Volume 7, Issue 4
The effect of solvents on spectroscopic properties of benzophenone is analyzed using DFT/6-311G method. The data on effect of solvents is used to estimate excited state dipole moment using the theoretically determined ground state dipole moment. The excited state dipole moment determined by different methods is compared and analyzed. The excited state dipole moment of benzophenone is found to be greater than its corresponding ground state counterpart and, ground and excited state dipole moments are almost perpendicular to each other. The study was aimed to study the energy, molecular structure, vibrational spectra, HOMO-LUMO analysis dipole moment. The wavelength and intensity absorption bands are both affected when a molecule is solvent environment. This is due to unequal perturbation of the ground state and exited state. The vibrational frequencies determined experimentally were compared with the DFT calculation on which was obtained theoretically employing the 6-311G(Dd,p) bassis set method for the optimized geometry of the benzophenone. Benzophenone, an aromatic ketone (diphenylketone), is an important compound in organic photochemistry and perfumery as well as in organic synthesis. It used as a consistent of synthetic perfumes and as starting material for manufacture of dyes, pesticides and drugs. It can act as optical filters or deactivate substrate molecules that have been excited by light for the protection polymer and organic substances. Benzophenone is produced by copper-catalyzed oxidation of diphenylmethane with air.
<Keywords: Benzophenone; Gaussian; Basis set; DFT; B3LYP; HOMO; LUMO
Background of study
Computational chemistry is fast developing branch of modern chemistry. Since the properties of substance depend on the properties of their molecules. Computational chemistry studies the properties of molecules to understand about the properties the substance. The more we can understand the properties of existing substances and be able to predict the properties of substances that have not yet been synthesized [1].
The term computational chemistry usually used when mathematical method is sufficiently well developed that it can be automated for implementation of computation. It is the application of chemical, mathematical and computational chemistry skills to the solution of interesting chemical problems. It uses computer to generate information such as properties of molecules or simulated experimental results. Very few aspects of chemistry can be computed exactly, but almost every aspects of chemistry has been described in a qualitative or approximate quantitative computation schemes [2].
The term theoretical chemistry may be defined as the mathematical description of chemistry. Currently there are two ways to approach theoretical chemistry problems. Computational theoretical Chemistry and non-computational theoretical chemistry. Computational theoretical chemistry is primarily concerned with the numerical computation of molecular electronic structure and molecular interactions and non-computational quantum chemistry deals with the formulation of analytical expression for the property of molecules and their reactions [3].
The most widely used software in computational chemistry is the Gaussian software. Gaussian uses the geometry optimization, frequency calculation, dipole moment, energy, molecular structure vibration spectra and transitional structure. The most important numerical techniques of Gaussian methods are ab initro, semiempirical and molecular mechanics [4].
Molecular geometry, optimized parameters and vibration frequencies performance of the computational methods for B3LYP at 6-31+G (d,p) and 6-311++G(d,p) basis sets compared. In addition, HOMO, LUMO and thermodynamic property have been used to elucidate regarding charge transfer within the molecules [5].
DFT calculation have been carried out B3P86/6-31+G(d,p) level of theory to explore and calculate more representative that can be related to the free radical scavenging ability of a series of 30 Schiff basses [6].
Study on solvent effect and estimation of dipole moments of an organic fluorophore, to analyze the solvent effects on absorption transition energy, fluorescence transition energy and stokes shift using different solvent polarity scale and DPHAPMC by different methods. The spectra properties are analyzed using Lippert and Mataga bulk solvent polarity parameters, Reichardt’s microscopic solvent polarity parameter and solvatochromic parameter proposed [7].
Using the solvent 2-chloro-5-nitrobenzyl alcohol, the density functional theory, the frequency calculation for 2C5NBA have been verified both experimentally and theoretically. Therefore, the present investigation was to study the vibration spectra of the molecule completely wave number accuracy. The DFT calculation have been performed to support wave number assignment, thermodynamic properties [8].
In this study, the effect of solvent on quantum mechanics in the properties of benzophenone and effects on the molecular geometry, optimization parameter, bond energy, vibration frequency, dipole moment is compared and the performance of the computational methods by PCM method. Benzophenone, an aromatic ketone (diphenylketone), is an important compound in organic photochemistry and perfumery as well as in organic synthesis. It used as a consistent of synthetic perfumes and as starting material for manufacture of dyes, pesticides and drugs. It can act as optical filters or deactivate substrate molecules that have been excited by light for the protection polymer and organic substances. Benzophenone is produced by copper-catalyzed oxidation of diphenylmethane with air (Figure 1).
Figure 1: Structure of benzophenone.
Physical and chemical properties of benzophenone
The physical and chemical properties of benzophenone are summarized in the Table 1 below.
| Physical state | Crystalline powder |
|---|---|
| Color | White flakes or crystals with rose like odor |
| Boiling point | 305-306°C |
| Melting point | 48-49°C |
| Flash point | 143°C |
| Solubility in water | Insoluble |
| Molecular formula | (C6H5)2CO |
| Molecular weight | 182.22 g/mol |
| Chemical stability | Stable under ordinary condition |
| Condition to avoid | Incompatible materials |
| Incompatibilities with other materials | Strong oxidizing agent |
| Specific gravity | 1.11 |
Table 1: Physical and Chemical properties of Benzophenone.
Objectives
General objectives: The general objectives of this study are to investigate the effect of different solvents on the quantum mechanical property of compounds.
Specific objectives: The specific objective of the study includes:
• To study the effect of solvent on Benzophenone by solvation methods.
• To know the polarity of the solvent, dipole moment, energy and frequency differences by using the PCM method.
• To calculate HOMO and LUMO levels using optimized geometrical parameters, dipole moment energy, frequency differences computed by density functional method (B3LYP) with 6-311G basis set.
Molecular structure
The optimized structure parameter of benzophenone calculated at DFT (B3LYP) level with the 6-311G(d,p) basis set are listed below in Table 2 in accordance with the atom numbering scheme given Figure 2.
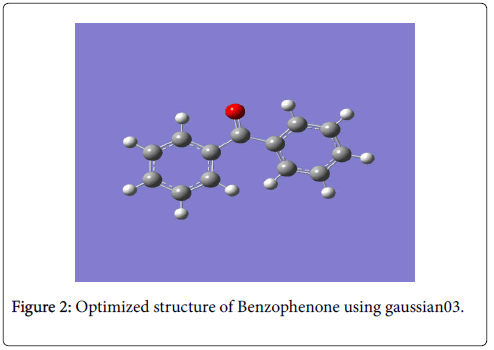
Figure 2: Optimized structure of Benzophenone using gaussian03.
| Solvent | Energy (in a.u) | Dipole moment (in D) | RMS Gradient norm (in a.u) | Spin | Basis set | Method | Charge | Jobs type |
|---|---|---|---|---|---|---|---|---|
| None | -576.60910813 | 3.0245 | 0.00002782 | Singlet | 6-311G | RB3LYP | 0 | Opt. |
| Water | -576.61090439 | 3.0518 | 0.00161712 | Singlet | 6-311G | RB3LYP | 0 | Opt. |
| Acetone | -576.61610837 | 4.4938 | 0.00173671 | Singlet | 6-311G | RB3LYP | 0 | Opt. |
| CCl4 | -576.62339926 | 4.0472 | 0.00001097 | Singlet | 6-311G | RB3LYP | 0 | Opt. |
Table 2: The calculated energy, dipole moment and RMS gradient for DFT/6-311G method of Benzophenone.
Solvent effect study
Benzophenone without solvent: The benzophenone without solvent have the calculated value of energy -576.60910813 a.u., the dipole moment of 3.0245D, and its RMS gradient norm of 0.0002782 a.u was calculated by B3LYP method in 6-311G basis sets.
Water as a solvent: Benzophenone was solvated with the water, the calculated value of energy -576.61090439 a.u, the dipole moment of 3.0518D, RMS gradient norm of 0.00161712 a.u. In this salvation, the dipole moment and the RMS gradient norm of the benzophenone was increased, but the energy decreased in the same method and basis sets, because of hydrogen bond. The water molecule contains partial positive and partial negative, that water is polar compound, as polarity increases dipole moment decrease but the energy increases.
Acetone: The calculation indicates that the energy value; -576.61610837 a.u, dipole moment of 4.4938D and its RMS gradient norm of 0.00173671 a.u. in this calculation, dipole moment and RMS gradient norm were increased, but the energy were also decreased in the same method (B3LYP) and 6-311G basis sets. The compound with solvent of Acetone was highest dipole moment. Because there were the same functional group and almost they have the same properties.
Carbon tetra chloride: The calculated value of benzophenone with CCl4 solvent; the energy, dipole moment and RMS gradient of -576.62339926 a.u, 4.0472D and 0.00001097 a.u respectively. In this calculated value the dipole moment was increased, but energy and RMS gradient norm were decreased.
Vibrational spectra
C-H vibration: In the aromatic compound the C-H stretching vibration of IR spectra observed at 3100-3000 cm-1. Most of the aromatic compound nearly three IR peaks in the regions 3080-3010 cm-1 due to the C-H stretching bond. In the present work, a sharp peak occurred at 3200 cm-1 by B3LYP/6-311G (d,p) method is due to the CH stretching vibration. The vibration peak at 3055 cm-1 in the experimental spectrum is assigned to C-H out of plane bending modes. Thus, the C-H vibration which was due to its high organic nature.
C-C vibrations: The ring C=C and C-C stretching vibrations, known as semicircle stretching occurs in the region 1625-1400 cm-1. The C=C stretching vibration for the title molecule benzophenone have four IR peaks at 1625, 1600, 1500 and 1400 cm-1 (6-311G (d,p) basis set). In the present work, most sharp peak occurred at 1600 cm-1 by B3LYP/ 6-311G methods. The experimental value for C=C stretching is observed at 1594 cm-1.
C-O vibration: The C=O stretching vibration of the benzophenone in the experimental results occurs in the region 1670-1640 cm-1. The experimental IR spectra sharp peak observed at 1634 cm-1, but the theoretical result occurs in the region 1625-1675 cm-1. From the present work, the most sharp peak calculated as 1650 cm-1 by B3LP/ 6-311G method. It was seen that the result of the B3LYP/6-31G method gave better resemblance to the experimental result in the IR analysis of benzophenone (Figures 3a-3c).
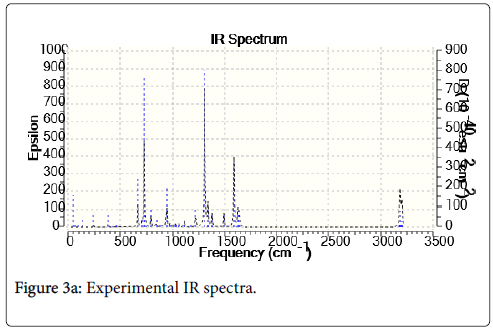
Figure 3a: Experimental IR spectra.
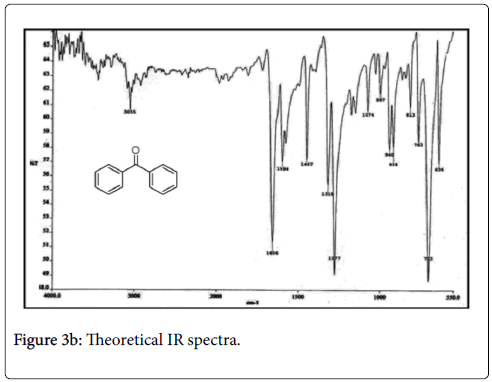
Figure 3b: Theoretical IR spectra.
Figure 3c: Display for vibration.
HOMO-LUMO analysis
The lowest lying unoccupied molecular orbitals and the highest occupied molecular orbitals are abbreviated as LUMO and HOMO, respectively. The HOMO containing electrons represents the ability to donate electrons, whereas LUMO have not electrons as an electron acceptor represents the ability to obtain an electron. There an energy gap between the HOMO and LUMO, and this energy gap determines the kinetic stability, chemical reactivity, optical polarizability of the molecule. The calculation indicates that the benzophenone has 48 occupied molecular orbitals and 49 unoccupied molecular orbital’s. The HOMO-LUMO energy gap of benzophenone was calculated using B3LYP/6-311G (d,p) basis sets. The HOMO-LUMO gap value of the studied compound is -0.26777 a.u, and -0.09269 a.u. respectively. The HOMO-LUMO orbitals picture (Figure 4).
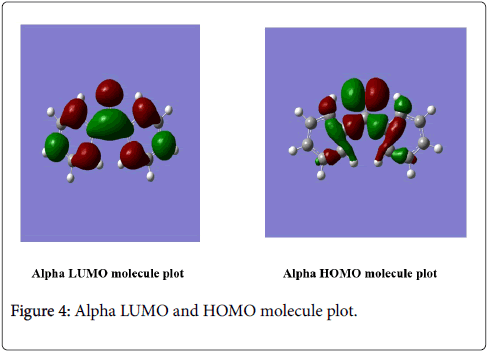
Figure 4: Display for vibration.
In this study, the vibrational frequencies, dipole moments, energy and IR spectra values for Benzophenone molecule were calculated, and the HOMO-LUMO analysis of the molecules were also performed by using DFT(B3LYP) method with 6-311G basis set. The dipole moment where found different solvents because of the solvent has different dielectric constant and the order of the dipole moment Acetone>CCl4>Water. In general, it’s possible to calculate the effect of solvent on the benzophenone in the energy, dipole moment, frequency differences and the HOMO-LUMO analysis of the organic compound using Gaussian 09w by PCM method (Table 3).
| Parameters | B3LYP |
|---|---|
| HOMO (a.u.) | -0.26575 |
| LUMO (a.u.) | -0.09033 |
| Energy (a.u.) | -576.60910813 |
| Dipole moment (in Debye) | 3.0245 |
Table 3: The calculated HOMO-LUMO energy and dipole moment using B3LYP methods with 6-311G basis set of Benzophenone.