Immunotherapy: Open Access
Open Access
ISSN: 2471-9552
ISSN: 2471-9552
Research Article - (2022)Volume 8, Issue 2
Pre-mRNA Processing Factor 31(PRPF31) is a key component of RNA splicing and a disease-causing gene of Retinitis Pigmentosa (RP). Previously, we found that the nonsense mutation R354X in PRPF31 induces RP in a Chinese RP family. In order to investigate the underlining molecular mechanisms of RP pathogenesis induced by this mutation, we generated cell lines stably expressing R354X mutant, Wild Type (WT) of PRPF31 and corresponding empty vector using HEK293T cells ,the resulting cell lines were used for Long non-coding RNA sequencing (LncRNA-sequencing). The results of LncRNA sequencing showed that, comparing to WT, R354X mutation changed the expression and splicing of coding and non-coding transcripts. Interestingly, in HEK293T and APRE-19 cells, inflammationassociated genes such as IFI6, OAS3and STAT3, enhanced their expression in response to the overexpression of WT PRPF31; however, in R354X mutation cells, those gene’s expression remained basal levels. Moreover, increased H2AFX expression and attenuated growth capacity were found in cells expressing R354X PRPF31 in HEK293T. In contrast with WT, R354X mutant showed varied splicing model on Di Hydro Folate Reductase (DHFR) in HEK293T. During IP analysis, we found that R354X mutant reduced its binding with CPSF1 and SORBS1 mRNAs in ARPE-19, and its binding with CTNNBL1 was also interfered in ARPE-19 cells. On the other hand, the R354X mutant also increased the level of transcripts read-through. Taken together, R354X mutation in PRPF31 affected cell survival, changed gene’s expression and splicing. Those findings indicate that inflammation and oxidation might contribute the pathogenesis of RP induced by the R354X mutation.
PRPF31; R354X mutation; Inflammation; DHFR; H2AFX
Retinitis Pigmentosa (RP) is an inherited ocular disorder, characterized by progressive degeneration of photoreceptors and abnormalities in retinal pigment epithelium [1]. In RP , the progressive death of Rod photoreceptors can cause early phenotype of RP, night blindness; then, induce the loss of cones photoreceptors ,and neuronal, glial and vascular remodeling, leading to the loss of vision [1,2]. Currently, more than 70 RP disease-causing genes are identified including hundreds of mutations [3]. The various disease-causing mutations in RP indicate complex molecular mechanisms during its pathogenesis including inflammation. The pro-inflammatory component is also a hallmark of the pathogenesis of RP [4-7]. Accumulated evidences showed that chronic and low-grade inflammatory processes play an important role in the RP progression [5-7]. An markedly elevated in the levels of proinflammatory cytokines detected in vitreous and aqueous humorn from RP patients [7].
As a constitutive component of spliceosomes, Pre-mRNAs Processing Factor 31(PRPF31) involves in the assembly and stabilization of U4/U6/U5 tri-snRNP [8-10]. Moreover, lossof- function analysis showed PRPF31 participates in snRNPs activation and splicing efficiency determination [11]. Insufficiency of PRPF31 can only impair the splicing of a subset of genes using patient-derived lymphocytes or siRNA-treated human organotypic retinal cultures [11]. Knocking PRPF31 out in zebrafish or mouse also severely affected the retinal-specific gene expression although mild disturbance for global transcriptome is also observed. PRPF31 mutation also causes RP [12,13]. Previously, we found that a nonsense mutation R354X in PRPF31 is a disease-causing mutation for a Chinese RP family [10]. This mutation can lead to an elevated PRPF31 protein expression [10]. In order to further understanding the potential molecular mechanisms of RP pathogenesis in this family, we performed a RNA sequencing analysis. We found that the changed expression for genes involving in inflammation in PRPF31 R354X mutant comparing to WT.
Cell culture and stable cell line establishment
HEK293T and human retinal pigment epithelial cell line (ARPE-19) were grown in Iscove’s modified Dulbecco’s medium (Invitrogen, U.S.A) with 10% fetal bovine serum, and 1% penicillin and streptomycin (Sigma-Aldrich, U.S.A.), and maintained in a humidified 5% CO2 incubator at 37 °C. Plasmids containing wild type PRPF31 and its R354X mutant were constructed with a GFP (Green Fluorescence Protein) merged at N terminus and a FLAG-tag merged at its C-terminus. These plasmids were used for the stable cell line establishment. Briefly, cells were seeded at a density of 2 × 105 cells per well in 6-well plates and cultured for 24 hours. Thereafter, each plasmid was transfected into cells using the Lipofectamine 3000 Transfection Reagent (Invitrogen, U.S.A.). The cells were incubated for 48 hours and then subjected to stable cell line screening by adding the puromycin to the medium. Cells stably expressing the wild type PRPF31 or PRPF31 R354X mutant were used for RNA extraction or other experiments.
RNA extraction and Reverse transcription
Total RNA’s were extracted from cells using TRIzol reagent (Invitrogen, U.S.A.) according to the manufacturer’s instruction. RNA integrity and concentration were assessed using RNA Nanodrop ND-2000 Spectrophotometer (Thermo, U.S.A.). Thereafter, the first stand of DNA synthesis was performed using the reverse transcription kit (Takara,Dalian) based on the manual instruct. The cDNA was used for qPCR or RT-PCR with the designated primer sets (Table 1).
| Name | Sequences(5’-3’) | Usage |
|---|---|---|
| IFI6-FP | CTGGTCTGCGATCCTGAATG | qPCR |
| IFI6-RP | AGAGGTTCTGGGAGCTGCTG | |
| STAT1-FP | CGGGTAGTTTCGCTTTCCTG | qPCR |
| STAT1-RP | GCTACGCACAGCACGTTAGG | |
| H2AFX-FP | GAGTACCTCACCGCTGAGATC | qPCR |
| H2AFX-RP | TGGCGCTGGTCTTCTTGGGCAG | |
| DHFR-FP1(50bp) | TCCAGAACATGGGCATCGGCAAG | PCR/qPCR |
| DHFR-RP1(50bp) | TGGAGGTTCCTTGAGTTCTCTGC | |
| DHFR-FP2(120bp) | TCCAGAAGTCTAGATGATGCCT | PCR/qPCR |
| DHFR-RP2(120bp) | TGCCTTTCTCCTCCTGGACATC | |
| PRPF31-GFP-FP-Xho1 | GC CTCGAG ATGTCTCTGGCAGATGAGCTCTTAGC | Plasmid construction |
| PRPF31-GFP-RP-EcoR1 | GC GAATTC GATTACAAGGACGACGATGACAAG GCTCAGGTGGACATAAGGCCACTCTTC | |
| PRPF31-GFP-RP-R354X-EcoR1 | GC GAATTC TCA GATTACAAGGACGACGATGACAAG CTTCTTCCGCTGTCCATCCAGG | |
| Beta-actin FP | ctc cat cct ggc ctc gct gt | qPCR |
| Beta-actin RP | gct gtc acc ttc acc gtt cc | |
| GAPDH FP | Agaaggctggggctcatttg | qPCR |
| GAPDH RP | aggggccatccacagtcttc | |
| CPSF1-FP | TCTACGTGCTGACCCTCATC | qPCR |
| CPSF1-RP | GCAGCTTCTCCGTGTACTTG | |
| SORBS1-FP | ACAGAGGGCAAGAAGGC | qPCR |
| SORBS1-RP | TCGGGAAGGTCAGAGGT |
Table 1: The cDNA was used for qPCR or RT-PCR with the designated primer sets.
LncRNA sequencing
The LncRNA sequencing services were provided by HUADA (BGI) Shenzhen, China. Cells stably expressing wild type PRPF31 and PRPF31 R354X were sent to BGI. Briefly, total RNA were extracted from the above cells with TRIzol reagent and further digested with DNase I. The resultant total RNA’s were then treated with RiboMinus Eukaryote Kit (Qiagen, USA) to remove the rRNA and fragmented using Ambion Fragmentation Solution (Thermo Fisher Scientific, USA). The fragmented RNAs were used for the cDNA synthesis. Following adenylation of DNA 3′-ends and hairpin loop structure ligation, the library fragments were purified to select desired cDNA fragments of 150-200 bp. The products were purified with the AMPure XP system and the library quality was assessed (Agilent Bioanalyzer 2100 system). The generated libraries were sequenced by BGI on an Illumina Hiseq 2500 platform, and 150-bp paired-end reads were produced.
Raw RNA sequence data analysis
The raw data of sequencing was filtered out step-by-step. We obtained the clean data through removing the adapter-polluted reads, low-quality reads and reads with more than 5% N bases accounting in the law data. Then, the resultant reads were mapped to the reference genome using HISAT2. The reference genomes and the annotation file were downloaded from the ENSEMBL database.
HTSeq was used for read count of each gene in each sample, and FPKM (Fragments Per Kilobase Million Mapped Reads) was then calculated to represent the expression level of genes in each sample, defined as FPKM=106 × F/ (NL × 10-3), where F is the number of fragments assigned to a certain gene in a certain sample, N is the total number of mapped reads in the certain sample, and L is the length of the certain gene. Thus, FPKM eliminates the influences of different transcript lengths and sequencing discrepancies on the calculation of expression. DESeq were used for differential expression analysis of two samples without replicates. A P-value could be assigned to each gene and adjusted by BH. Genes with q<0.05 and |log2 ratio|>1 were identified as differentially expressed genes.
Quantitative real-time polymerase chain reaction
qRT-PCR was performed using the TB Green Premix Ex Taq (Tli RNaseH Plus) (TAKARA, Dalian, China) according to the manufacturer’s instructions. Each reaction contained 5 μl TBGreen Premix Ex Taq qPCR Master Mix (Promega), 5 μl cDNA sample, nuclease-free water and 0.5 μl primers (10 μM). All amplified products ranged from 100 to 200 bp in size. The plates were run on an Applied Biosystems 7500 fast Real Time PCR machine. The cycling program consisted of hot-start activation at 95°C for 5 min, followed by 45 cycles of denaturation at 95°C for 10 sec, annealing/ extension at 60°C for 30 sec and denaturation 95°C for 1 min. Following amplification, a melt-curve analysis was performed from 65 to 95°C with 0.5°C increments every 10 sec. Each sample was run in triplicate, and the average quantification cycle (Cq) value was determined. Control reactions were run with water instead of template for each primer pair to check for primer-dimmers and reagent contamination. Normalised gene expression values (against GAPDH or beta-actin) were obtained using the ΔΔCT method. All primer details are shown in Table 1.
Western blotting
Cells transfected with designated plasmids were lysated in 1 × NP40 Lysis buffer containing the complete protease inhibitor (Roche, USA). Then the lysates were centrifuged at 4oC, 15000 g for 15 min. The supernatants were used for western blotting analysis. Proteins (ranging from 20 to 50 µg) from each sample were electrophoresed on a 10% Bis-Tris Criterion XT Precast gel (Bio-Rad, U.S.A), then blotted on 0.2 nitrocellulose membrane using a transfer system (Bio-Rad). The protein blot was blocked by exposure to 5% non-fat milk TBST solution at room temperature for 1 hour. Nitrocellulose filters were then incubated overnight at 4°C with the primary antibodies: anti-STAT3 (Ab226942, abcam), anti-IFI6 (G1P3 Antibody (H-84)) (IC162417; X-Y biotechnology, China), anti-Flag (SAB4200071, Sigma), Anti-H2AFX biovision, anti-CTNNBL1 (ab95170) and secondary antibodies. The GAPDH (ab8245, abcam) was used as loading control.
Examination of cell viability
Cell viability was determined using an MTT assay (Sigma-Aldrich, USA). Cells stably expressing PRPF31 WT or R354X were cultured in 24-well plates at a concentration of 2 × 105 and allowed to grow to 24, 48 and 72 hours. Following growth for different time intervals, 100 µl MTT (0.5 mg/ml) was added into the cells and the mixture was incubated for 4 hours at 37°C. Subsequently, the supernatant was removed and dimethyl sulfoxide (Sigma-Aldrich, USA) was used to dissolve the resultant formazan crystals. The absorbance was read at 570 nm (EL309 Automated Microplate Reader; Bio-Tek Instruments). Six wells were measured for each group and the experiment was repeated three times.
RNA immunoprecipitation (RIP)
RIP assay was carried out in ARPE-19 cells using Magna RIP Kit (Millipore, USA) following the manufacturer’s instructions. In brief, magnetic beads pre-coated with 5 mg normal antibodies against Flag (Sigma, USA) or mouse IgG (Millipore) were incubated with sufficient cell lysates (more than 2 × 107 cells per sample) at 4°C overnight. And the beads containing immunoprecipitated RNA-protein complex were either used for WB analysis to test the CTNNBL1 binding, pulldown efficiency or treated with proteinase K to remove proteins for qPCR analysis. Then interested RNA’s were purified by TRIzol methods (Thermo Fisher Scientific) and detected by RT-qPCR with the normalization to input (fold change was calculated for comparison).
1. LncRNA sequencing data was qualified and large number of useful information was discovered
As our previous paper showed, R354X mutation in PRPF31 could induce development of Retinitis Pigmentosa (RP). In order to further understand its molecular mechanisms, we generated three lines, which can stably express R354X mutation or WT of PRPF31 or Negative Control (NC) using HEK293T cells. After that, we used those cell lines for LncRNA sequencing with Illumina Hiseq platform with a read length 150 bp (Ruohe, China). The rRNA but not the mRNA's were removed from the total RNA’s samples before cDNA synthesis. Thus, we can analyze both mRNA’s and non-coding RNA’s in samples. The sequencing data from all samples were qualified and the effective reads were all close to 99.6% (Figure 1A).The number of clean reads (about 67.7 M), clean bases (more than 10 Gb), Q30 (about 94.5%) and Q20 (about 97.5%) are almost the same. The GC content has no significant difference in all three samples.
2. Differential expression and splicing in transcripts between groups expressing R354X mutant and WT of PRPF31
The number of total reads and total mapped reads were almost the same between these two groups (Figure 1A). However, we can easily notice the significant difference between groups expressing R354X mutant and WT of PPRF31 when we compared the mapped reads number between spliced (18.54%, 19.26% for mutant and WT, respectively) and non-spliced (29.23%, 27.86% for mutant and WT, respectively) (Figure 1A). In contrast, over-expressing WT or R354X mutant of PRPF31 had higher exon read number than no transfected cells (NC) (exon read for WT 22%, R354X 23%, NC 18.4%); but decreased the number of Intron reads (59.7% (NC), 56.4% (WT) and 53.8% R354X mutation, respectively) (Figure 1B). Venn diagram illustrated common differentially expressed genes in three groups in coding RNA’s (left) and non-coding RNA’s (right) (Figure 1C). The detailed information for total significantly differential mRNA’s and non-coding RNA’s. Heatmaps generated from differentially expressed non-coding RNA’s and mRNA’s showed R354X mutation could affect large numbers of transcripts expression (Figure 2A). Moreover, Gene ontology with KEGG analysis showed that genes involving in infection, immunity and metabolisms were significantly changed in R354X mutated PRPF31 stable cell lines (Figure 2B). A similar result was also observed in non-coding RNA’s and microRNA’s. R354X mutation in PRPF31 also decreased the number in RNA read through (Figure 2C) and changed the expression of many genes (Figures 2D and E), comparing to WT.

Figure 1: Statistical analysis of LncRNA data on mRNAs.
A: Statistical analysis that reads mapped to reference genome; B: Statistical analysis of transcript structure in three different cell lines; C: Venn diagram
illustrating common differentially expressed genes in three groups in coding RNAs (left) and non-coding RNAs (right). NC represent cells stably
expressing empty vector; WT represent cells stably expressing wild type of PRPF31; R354X represent cell stably expressing R354X mutated PRPF31.


Figure 2: Statistical analysis for mRNA data from LncRNA sequencing.
A: Statistics of pathways, enrichment in the KEGG database for neighbouring genes of predicted mRNAs. Pathways had q values <0.05 and were
considered as significantly enriched pathways; B: Hierarchical heat map displaying transformed expression values (Log2FPKM) for significantly
differentially expressed mRNA’s. Red shows up-regulation and green shows down-regulation; C: Read through gene number in WT and R345X;
D: Number of differential regulation genes (WT to R354X), fold change >2 or <-2, p<0.05; E: Volcano plot of mRNAs between R354X and WTs.
2.0- fold-change up-and down-regulation, P value<0.05.
3. R354X mutation in PRPF31 decreases the alternative splicing of RI and A3SS
PRPF31 is a key protein in the regulation of RNA splicing. We thus analyzed the LncRNA sequencing data in order to observe whether alternative splicing events were affected after R354X mutation. Interestingly, R354X mutation leads to the reduction of RNA splicing event, especially in types of RI and A3SS, which were almost removed from R354X mutation PRPF31 cells (Figures 3A and B). Other three types of splicing were also affected partially comparing to WT.

Figure 3: Statistical analysis of differentially alternative splicing.
A: R354X PRPF31comparing to empty vector; B: WT PRPF31 to empty vector.
Note: SE: Skipped Exons; A3SS, A5SS: Alternative3'/5' Splice Sites; MXE: Mutually Exclusive Exons; RI: Retained Introns.
4. qPCR and Western blotting were used to confirm the selected differential expression mRNA’s
We chose two down-regulated (STAT1, IFI6) and one up-regulated (H2AFX) genes for qPCR and Western Blotting (WB) analysis in HEK293T or ARPE-19 cells. The results showed that RNAs expression for STAT3 in R354X mutant significantly down- regulated comparing to WT, consisting with the results of LncRNA sequencing. On the other hand, H2AFX was significantly enhanced in R354X PRPF31 expressed cells in contrast with WT, for both mRNA and protein, which is also consistent with the LncRNA sequencing. Because the enhanced expression of H2AFX can induce cell apoptosis, we then checked the cell survival via MTT analysis. We found that cells stably expressing R354X PRPF31 proliferate more slowly than cells expressing WT PRPF31. The protein expression of WT and R354X of PRPF31 was showed as in Figure 4. This result indicates that R354X mutation in PRPF31 damages the capacity of cell growth.

Figure 4: The expression levels of STAT3, IFI6 and H2AFX.
Expression levels were normalized to beta-actin. Data are the means ± S.D of three independent experiments. **P<0.01,*p<0.05, one-way analysis
of variance followed by Dunnett’s multiple comparison test or Student’s t-test was performed. ET, represents cells stably expressing empty vector.
5. R354X mutation in PRPF31 changed the alternative splicing of DHFR
Based on the results of LncRNA sequencing, we found that the MXE type of splicing at the second and forth exon in DHFR was strongly affected in R354X mutant HEK293T cells. So we designed two primer sets (FP1/RP1, FP2/RP2) to detect this alternative splicing event using RT-PCR. In contrast to WT group, we observed different sizes of PCR products in R354X mutated PRPF31 via same primer set in HEK293T. Quantifying the above PCR products with qPCR showed differential expression level of those alternative splicing products between R354X mutant and WT (Figure 5), showing a consistent result with the sequencing data.

Figure 5: Confirmation of alternative splicing on DHFR in cells expressing R354X mutant.
A: alternative splicing result between WT and R354X on DHFR gene from lncRNA sequencing; B: schematic diagram showed the alternative
splicing exons and locations of primers used to confirm the splicing; C: multi-alignment of three alternative transcripts in DHFR genes showed
differential splicing sequence; D: multi-alignment of three alternative transcripts in DHFR genes showed differential splicing sequence; E: RT-PCR
analyzing the alternative splicing exons within cells stably expressing WT or R354X PRPF31; F: qPCR analysis to the alternative splicing exons with
the same cell lines as in E. data was expressed as mean relative mRNA expression ± S.D. 
6. Immunoprecipitation further showed that R354X mutant affected the splicing capacity of PRPF31
CPSF1 and SORBS1 genes are the substrates of PRPF31-mediated splicing. We thus compared the binding level of WT and R354X mutant to those two RNA’s via Immune Precipitation (IP) in APRE- 19 via anti-Flag antibody. As expected, R354X mutant showed a reduced binding capacity to the above two RNA’s, compared to WT. The IP efficiency was confirmed by checking the pull-downed WT and R354X mutant via WB. Also, we found that CTNNBL1, which was confirmed to be a binding protein during splicing process (Figure 6).

Figure 6: R354X mutant affected inflammation-associated protein’s expression and splicing in ARPE-19 cells.
A: ARPE-19 cells stably expressed the WT and R354X mutant of PRPF31, Western blot analysis to the expression of STAT1, IFI6 and H2AXF,
GAPHD as a loading control. B, C, RNA IP and co-IP analysis; B: qPCR analysis for the pulldown RNAs with designated primers; C: Using Flag
and IgG antibody to IP and Proteins or RNAs pull-downed was used to WB directly or RNA extraction with TRIzol reagent.
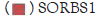
Mutations in PRPF31 induce the pathogenesis of autosomal dominant retinitis pigmentosa. Recently research showed knockout PRPF31 in zebrafish affects the differentiation and viability of retinal progenitor cells [6]. Previously, we found that several mutations in PRPF31 induced the pathogenesis of RP [10]. Of those mutations, R354X mutation showed a more stable protein comparing to the WT although C-terminus is deleted. In order to test its underlining molecular mechanisms, we generated three cell lines stably expressing Empty vector, R354X PRPF31, WT PRPF31 via HEK293T and then did LncRNA sequencing with those cell lines. We found that R354X mutation in PRPF31 affected RNA splicing and expression of certain transcripts. Interestingly, we uncovered that over-expressed PRPF31 in HEK293T strongly activated the expression of inflammation-associated genes such as IFI6. However, in cells stably expressing R354X mutant, IFI6 expression could not be induced. Moreover, the key transcriptional factor for expression of inflammation genes, STAT3 was also activated in WT but not in R354X mutant. Because inflammation, autophagy and oxidative stress are very important factors during the pathogenesis of RP [6,7,14], mutation at the R354 in PRPF31 might prevent the ability of PRPF31’s response to various infections and lead to the damage of infected retinal tissues.
We also noticed that H2AFX expression significantly increased in R354X mutant comparing to WT. H2AFX is a marker of DNA damage [15]. Previous findings showed that knockout PRPF31 in zebrafish, Retinal Progenitor Cells (RPCs) displayed significant mitotic arrest and DNA damage, which was rescued by the wild type human PRPF31 [12,13,16]. Further analysis showed knockout PRPF31 caused the skipping of exons with a weak 5’ splicing site. The genes involve in the DNA repair and mitotic progression are affected [12,13,16]. Our current finding is in line with this. R354X mutation in PRPF31 affects its splicing capacity, and in turn prevents the biological roles of DNA repair associated genes.
R354X mutant not only alters gene expression, but also undermines its splicing capacity (Figures 5 and 6). DHFR is an important enzyme required to maintain bacterial growth, and hence inhibitors of DHFR have been proven as effective agents for treating bacterial infections [17]. CPSF1 was reported as a regulator of retinal ganglion cell development, therefore, un-proper splicing for this gene, might affect ocular functions.
Taken together, we found R354X mutation in PRPF31 changed its splicing capacity and exposed the retinal tissue to be vulnerable after infection.
Mutations in PRPF31 induce the pathogenesis of autosomal dominant retinitis pigmentosa. Recently research showed knockout PRPF31 in zebrafish affects the differentiation and viability of retinal progenitor cells. Previously, we found that several mutations in PRPF31 induced the pathogenesis of RP. Of those mutations, R354X mutation showed a more stable protein comparing to the WT although C-terminus is deleted. In order to test its underlining molecular mechanisms, we generated three cell lines stably expressing Empty vector, R354X PRPF31, WT PRPF31 via HEK293T and then did LncRNA sequencing with those cell lines. We found that R354X mutation in PRPF31 affected RNA splicing and expression of certain transcripts.
Ethical statement
The study was approved by the Ethics Committee of the Joint Shantou International Eye Center (JSIEC), Shantou University & the Chinese University of Hong Kong, Shantou, Guangdong province, China conducted in accordance to the Declaration of Helsinki.
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google scholar] [PubMed].
[Crossref] [Google scholar].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar] [PubMed].
[Crossref] [Google Scholar].
Citation: Xiao X, Deng F, Huang S (2022) R354X Mutation in PRPF31 affects its Capacity in Inflammation Response and Splicing. Immunotherapy (Los Angel). 08:190.
Received: 11-Mar-2022, Manuscript No. IMT-22-16154; Editor assigned: 15-Mar-2022, Pre QC No. IMT-22-16154 (PQ); Reviewed: 30-Mar-2022, QC No. IMT-22-16154; Revised: 04-Apr-2022, Manuscript No. IMT-22-16154 (R); Published: 12-Apr-2022 , DOI: 10.35248/2471-9552.22.08.190
Copyright: © 2022 Xiao X, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.