Cell & Developmental Biology
Open Access
ISSN: 2168-9296
ISSN: 2168-9296
Research Article - (2014) Volume 3, Issue 1
Ral, a Ras-like GTPase, is an essential component of Ras-driven oncogenesis that supports tumor initiation and progression. We explored the role of Ral in the polarity organization of epithelial tissue in Drosophila. We found that Ral is dispensable for the polarization of proliferative epithelial tissue, and that Ral is required for the maintenance of apical-basal polarity of post-mitotic epithelial cells during tissue remodeling. More precisely, lack of Ral activity results in loss of cortical apical aPKC and lateral Lgl, and in apical spreading and accumulation of Armadillo/betacatenin. Our analysis demonstrates that Ral regulates polarity organization by acting as a negative regulator of JNK and p38 MAPK signaling. Ral controls aPKC apical localization by down-regulating JNK and controls Armadillo localization at adherens junction by down-regulating p38 MAPK signaling, whereas Lgl membrane localization depends on both pathways. Finally, in the absence of Ral function aPKC becomes dispensable for Lgl basolateral localization and Armadillo is recruited to the cell membrane independently of DE-Cadherin and Bazooka localization. This suggests that additional mechanisms control Lgl and Arm distribution in polarized epithelia.
<Ral, a Ras-like GTPase, is an essential component of Ras-driven oncogenesis supporting tumor initiation and progression [1,2]. Moreover, the important role of Ral in many cellular events affected in cancer cells, such as polarized exocytosis, apoptosis, autophagy and migration, was recently demonstrated [3-6]. Apical-basal (AB) polarity is a fundamental to epithelial organization and several human pathologies, including cancers are associated with defects in epithelial polarity. Yet the role of Ral in epithelial cell polarity is only partially deciphered [7-10]. In particular, the analysis of the Ral polarity function in Drosophila developing tissue has been conducted using overexpression of dominant active or negative Ral forms. This analysis has demonstrated a Ral function in AB polarization of dividing neuroblasts [7].
Epithelial cells comprise at least four distinct cortical domains: an apical domain, an apical-lateral junction (the adherens junction in Drosophila), a lateral domain and a basal domain [11,12]. AB polarity is achieved by the concerted action of several conserved protein complexes. The Crumbs complex (Crumbs/Pals/Patj) and the Par complex (aPKC/Par6/Par3) are associated with apical domain. Adherens Junction (AJ) complex contains DE-Cadherin (DE-Cad), Armadillo/β-catenin (Arm) and a-catenin. Basolateral domain contains the Scribble complex (Scrib/Dlg/Lgl) and the recently identified Yurt/Coracle group (Yurt/Cora/NaK-ATPase/Nrx-IV). Maintenance of AB polarity depends in part on antagonistic interactions between the polarity complexes, which define distinct cortical domains and the position of the adherens junctions [13-20].
Follicular epithelium of Drosophila ovary is an important model system to analyze the mechanisms underlying establishment and maintenance of epithelial cell polarity in vivo. Ovarian follicle or egg chamber is the structural and functional unit of insect ovary. When cell divisions end each egg chamber consists of 16 germ line cells (15 nurse cells and oocyte)surrounded by a monolayer of approximately 850-900 somatic epithelial cells called Follicle Cells (FC). Egg chambers are formed in the anterior tip of the ovariole, in a region called germarium. They bud from the germarium (stage 2) and continue to move posterior through the ovariole as oogenesis proceeds. Initial polarization of FC takes place within the germarium and in stage 1 egg chambers [21]. FC stop proliferating at stage 6/7, enter an endocycle and begin to differentiate. By stage 8, they are fully differentiated along the anterior-posterior axis of the oocyte, forming a morphologically uniform cuboidal epithelium, around the germline tissue [22]. Follicular epithelium undergoes tissue remodeling including both extensive cell shape changes and directed cell migration during stage 9. These morphogenetic rearrangements result in the characteristic organization and distribution of cells in egg chambers at stage 10A. Majority of FC cover the posterior-positioned oocyte and take on a distinctive, highly columnar morphology and eventually synthesize the eggshell. Concurrently, the most anterior FC forms a so-called squamous epithelium, which covers the nurse cells [23-25]. Hence, ovarian follicular epithelium development permits to distinguish and analyze gene functions underlying polarity maintenance during either mitotic divisions or epithelial tissue remodeling.
Using a newly generating Ral null loss-of-function allele, we show that Ral is not required for the polarization of dividing epithelial cells, but that it is essential to maintain the polarity of post-mitotic epithelial cells during tissue remodeling. We demonstrate that the absence of Ral activity causes loss of apical aPKC, loss of lateral Lgl and Lgl cytoplasmic mislocalization, and Arm apical spreading and accumulation. Our genetic analysis indicates that Ral controls epithelial polarity by down-regulation of the JNK and p38 MAPK pathway activities. Finally, our analysis of the Ral loss of function suggests additional mechanisms for the maintenance of polarity in post-mitotic epithelial cells since in the absence of Ral function, aPKC becomes dispensable for Lgl basolateral localization and Arm is recruited to the cell membrane independently of DE-Cad localization.
Fly stocks and genetics
Mutations and constructs are described in Fly Base (http://flybase.bio.indiana.edu). All mutant alleles used in the study are either null or strong loss of function mutations. The following mutant alleles were used: msn172; bsk1; mpk21 (p38a); 14-3-3εj2B10; mekk1UR36; fafBX4; wnd2; sec5E13; Ral35d; pucE69. The following UAS transgenes were used in this study: msnDN; msnWT; DjunDN; wndKDandhiwWT; p38WT; RalWT. Mutants were kindly provided by J. Fischer (faf), A. DiAntonio (wnd and hiw), H. Inoue (mekk1), Y. Rao (msnDN) and T. Adachi-Yamada (p38WT). Other mutants were provided from Bloomington and VDRC stock centers.
Clonal analyses in follicular epithelium were performed with FLP/FRT system using nuclear GFP as marker of control cells [26]. T155-Gal4 driver was used both for clone induction and transgene overexpression in FC. Crosses for clone induction were done at 27°C. Clones were analyzed in the follicular epithelium of the egg chambers from stage 2 till stage 10 in Ral70a, FRT101/Ubi-GFP, FRT101; T155-Gal4, UAS-Flp flies(Ral polarity phenotypes); in Ral70a, FRT101/Ubi-GFP, FRT101; mutX/+; T155-Gal4, UAS-Flp(2nd chromosome) or Ral70a, FRT101/Ubi-GFP, FRT101; mutX/T155-Gal4, UAS-Flp(3rd chromosome) (modulations of Ral-induced polarity phenotypes). msn172 and mpk21 control clones were induced using hsFlp and 2 hours 37°C heat shocks on two consecutive days during the second instar. Clones were analyzed in y, w, hsFlp/+; msn172, FRT80B/Ubi-GFP, FRT80B and in y, w, hsFlp/+; FRT 82B, mpk21/FRT82B, Ubi-GFP (respectively). Ral germline clones were generated by the FRT/FLP-ovoD1 system [27] using hs FLP and 2 hours 37°C heat shocks on two consecutive days during the second instar.
Immunochistochemistry
Egg chambers were fixed and stained as previously described [28].Primary antibodies were: mouse anti-Grk 1D12 (1:10; Developmental Studies Hybridoma Bank, DSHB), goat anti-Staffen (1:50, provided by A. Guichet), rat anti-DE-cadherin DCAD2 (1:20; DSHB), mouse anti-Arm N27A1 (1:50; DSHB), mouse anti-Crumbs Cq4 (1:50; DSHB), rabbit anti-dPatj (1:500, provided by M. Bhat), rabbit anti-PKCζ C20 (1:500;Santa Cruz Biotechnology, Inc.), rabbit anti-Baz (1:500, [29], rabbit anti-Lgl (1:200, [13], mouse anti-Dlg 4F3 (1:500; DSHB), rabbit anti-Sdt (1:100, provided by A. Wodarz), rabbit anti-p-JNK (1:200, Promega). Secondary antibodies conjugated to Cy3 (Interchim, Jackson Immuno Reseach) were used at 1:100. Imaging was performed using Leica SP2 and Zeiss LSM780 confocal microscopes with 40x oil lenses. Images were processed using Image and Adobe Photoshop.
Biochemistry
Protein extracts from whole embryos and 1st instar larvae were prepared as previously described [4]. 75 µg of proteins/lane were separated in 12%-acrylamide SDS-PAGE gels, transferred on nitro-cellulose membranes and incubated overnight at 4°C using primary antibodies: anti-drosophila-Ral (1:500), mouse anti-p-JNK (1:250, Santa Cruz Biotechnology), rabbit anti-ERK1/2 (1:1000, Sigma). A polyclonal antibody against Drosophila Ral protein was raised in guinea pig by injection of the entire Ral protein (produced in bacteria as GST-fusion) (Eurogentec, Belgium).
Ral is implicated in the maintenance of apical-basal polarity in post-mitotic epithelial cells
Two classes of Ral alleles, lethal or viable but displaying a loss-of-bristle phenotype, have been obtained by P-excision mutagenesis [4]. Viable Ral mutants are hypomorphic alleles of Ral. Ral70a is a lethal allele of Ral, and Ral70a homozygotes die as embryos or 1st instar larvae. More than 2.7 kb of DNA are deleted from the Ral locus (data not shown) and the amount of Ral protein detected in Ral70ahomozygotes by Western blot analysis is considerably reduced. The amount of Ral protein observed in Ral70aembryos being 2 fold-lower than the one observed in Ral35dembryos (Figure 1A). The viability of Ral70a embryos is rescued by expression of UAS-Ral under control of ubiquitous Gal4 driver. These data show that Ral70a is a Ral null allele.
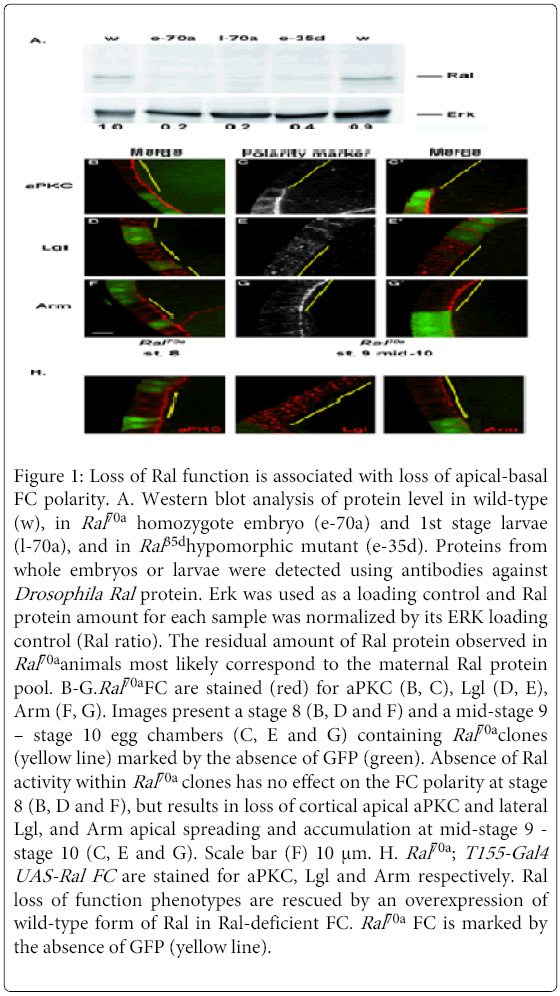
Figure 1: Loss of Ral function is associated with loss of apical-basal FC polarity. A. Western blot analysis of protein level in wild-type (w), in Ral70a homozygote embryo (e-70a) and 1st stage larvae (l-70a), and in Ral 35dhypomorphic mutant (e-35d). Proteins from whole embryos or larvae were detected using antibodies against Drosophila Ral protein. Erk was used as a loading control and Ral protein amount for each sample was normalized by its ERK loading control (Ral ratio). The residual amount of Ral protein observed in Ral70a animals most likely correspond to the maternal Ral protein pool. B-G.Ral70a FC are stained (red) for aPKC (B, C), Lgl (D, E), Arm (F, G). Images present a stage 8 (B, D and F) and a mid-stage 9 – stage 10 egg chambers (C, E and G) containing Ral70a clones (yellow line) marked by the absence of GFP (green). Absence of Ral activity within Ral70a clones has no effect on the FC polarity at stage 8 (B, D and F), but results in loss of cortical apical aPKC and lateral Lgl, and Arm apical spreading and accumulation at mid-stage 9 - stage 10 (C, E and G). Scale bar (F) 10 μm. H. Ral70a ; T155-Gal4 UAS-Ral FC are stained for aPKC, Lgl and Arm respectively. Ral loss of function phenotypes are rescued by an overexpression of wild-type form of Ral in Ral-deficient FC. Ral70a FC is marked by the absence of GFP (yellow line).
In addition to the loss-of-bristle phenotype, several Ral hypomorphic alleles displayed a female sterility phenotype. Female sterility observed in Ral hypomorphic mutants suggested that reduced Ral activity might lead to defects in oogenesis. We first analyzed whether loss of Ral function affected germline development. Analysis of Ral70a germline clones revealed that the absence of Ral activity did not affect the localization of either Oskar or Gurken proteins or the oocyte morphology (data not shown). Thus, Ral signaling is dispensable for polarization and morphogenesis of the oocyte. We therefore undertook an analysis of Ral function in FC. We performed our analysis in egg chambers, from stage 2, when egg chambers have already segregated from the germarium and initial specification of the apical membrane is finished [21], until stage 10B, after which nurse cells and FC undergo terminal apoptosis and eggshell secretion [24]. We examined polarity organization in Ral70aFC by studying the localization of Bazooka (Baz) (Par3 in mammals) and aPKC proteins (Par apical complex), Crumbs (Crb), Stardust (Sdt/Pals1) and DPatj proteins (Crumbs apical complex), Dlg and Lgl proteins (Scribble lateral complex), DE-Cad and Arm (β-catenin in mammals) (AJ complex).
No polarity defects were observed before middle stage 9 either in small (1-3 cells) or in big (more than 10 cells) Ral70a clones (Figures 1B, 1D and 1F). However we observed specific polarity defects in Ral70a FC at the late stages of oogenesis (mid-stage 9 - stage 10, Figure 1I). Whereas Baz (n=12/12 clones), DE-Cad (n=11/11 clones), Dlg (n=7/7 clones), Crumbs (n=9/11 clones), DPatj (n=7/7clones), and Sdt (n= 3/3 clones) were correctly localized (Figures S1A-F), the distributions of aPKC, Lgl and Arm were altered by Ral loss of function (Figures 1C, 1E and 1G). The absence of Ral activity compromised aPKC apical localization (Figure 1C, n=19/21 clones), Lgl lateral localization, resulting in its cytoplasmic mislocalization (Figure 1E, n=13/14 clones), and induced Arm apical spreading and accumulation (Figure 1G, n=14/15 clones). The co-staining of Arm either with Baz or with DE-Cad demonstrated that in Ral-deficient FC Arm apical spreading and accumulation is observed whereas Baz and DE-Cad are correctly localized at AJ (Figures S1G and H).
Neither apoptosis nor hyperproliferation of FC were observed in Ral70a clones or in surrounding control cells (Figure 1). Ral70a polarity phenotypes were cell autonomous and were rescued by the expression of wild-type form of Ral in Ral70a FC (n=16/20, n=13/13 and n=10/10 clones for aPKC, Lgl and Arm, respectively) (Figure 1H). Thus, Ral loss of function altered polarity organization of the FC that have stopped proliferating and come through morphogenetic rearrangement.
Ral controls polarity organization of FC by regulating JNK and p38 MAPK activation
Previously we showed that Ral induced apoptosis during sensory organ development by acting as an upstream regulator of the JNK and the p38 MAPK pathways [4]. Accordingly, we observed that lack of Ral activity in Ral70a follicle cells (n=8/9 clones) or decreasing Ral activity in Ral35d hypomorphic mutant FC resulted in elevated level of phosphorylated JNK (Figure 2A). The strong increasing of phospho-JNK level was also observed in homozygote Ral70a embryos and 1st stage larvae (Figure 2B). We supposed that Ral might regulate AB polarity in FC cells by down regulating JNK and/or p38 MAPK signaling pathways. The localization of aPKC, Lgl and Arm were not affected inmsn172 or mpk21 (encodes p38a MAPK), clones as well as in T1555-Gal4>UAS-bskRNAi FC (data not shown). We therefore explored genetic interactions between Ral and the components of JNK and p38 MAP kinase pathways using either loss-of-function mutations and overexpression of Dominant Negative (DN) alleles to reduce signaling or overexpression of Wild-type (WT) forms to promote these pathways (Figures 2C-2N and S2A-I).
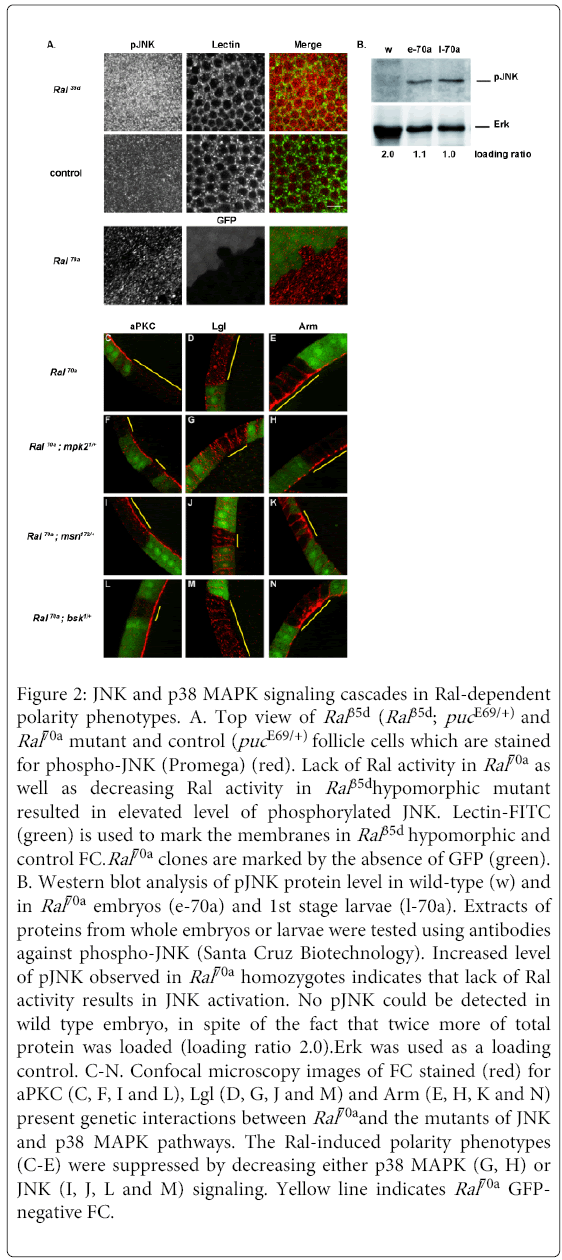
Figure 2: JNK and p38 MAPK signaling cascades in Ral-dependent polarity phenotypes. A. Top view of Ral 35d (Ral 35d; puc E69/+) and Ral70a mutant and control (puc E69/+) follicle cells which are stained for phospho-JNK (Promega) (red). Lack of Ral activity in Ral70a as well as decreasing Ral activity in Ral 35dhypomorphic mutant resulted in elevated level of phosphorylated JNK. Lectin-FITC (green) is used to mark the membranes in Ral 35d hypomorphic and control FC.Ral70a clones are marked by the absence of GFP (green). B. Western blot analysis of pJNK protein level in wild-type (w) and in Ral70a embryos (e-70a) and 1st stage larvae (l-70a). Extracts of proteins from whole embryos or larvae were tested using antibodies against phospho-JNK (Santa Cruz Biotechnology). Increased level of pJNK observed in Ral70a homozygotes indicates that lack of Ral activity results in JNK activation. No pJNK could be detected in wild type embryo, in spite of the fact that twice more of total protein was loaded (loading ratio 2.0).Erk was used as a loading control. C-N. Confocal microscopy images of FC stained (red) for aPKC (C, F, I and L), Lgl (D, G, J and M) and Arm (E, H, K and N) present genetic interactions between Ral70a and the mutants of JNK and p38 MAPK pathways. The Ral-induced polarity phenotypes (C-E) were suppressed by decreasing either p38 MAPK (G, H) or JNK (I, J, L and M) signaling. Yellow line indicates Ral70a GFPnegative FC.
We observed that reduction of the activities of Bsk, the Jun N-terminal Kinase (JNK) or Msn, the MAP4K4 orthologous of vertebrate HGK were sufficient to rescue the aPKC (n=12/15 and n=12/12 clones, respectively) and Lgl (n=10/10 and n=11/12 clones, respectively) polarity defects observed in Ral70a FC (Figure 2C and2D) and wild-type membrane localization of aPKC and Lgl was restored in Ral70a clones induced in bsk1/+ FC (Figures 2L and 2M) as well as in msn172/+ (Figures 2I and 2J) or in T155-Gal4>UAS-msnDN FC (Figures S2A and B). Accordingly, both phenotypes were rescued by down-regulation of JNK targets. Surprisingly, overexpression of the dominant negative allele of JNK transcriptional factor Djun (UAS-Djunbzip) restored wild-type membrane localization of Lgl (n=13/13 clones) but didn’t rescue apical aPKC localization (n=8/8 clones) (Figure S2E and F). To further confirm the role of JNK signaling in the aPKC polarity defect we choose one of the JNK targets, the 14-3-3ε protein, which had been found among the modifiers of Ral-apoptotic phenotype in our gain-of-function screen (data not shown). We observed that the decrease of the 14-3-3ε protein level rescued Ral-induced aPKC phenotype (n=17/23 clones) and wild-type membrane aPKC localization was observed in Ral70a; 14-3-3εj2B10/+ FC (Figure S2G). Thus, loss of apical aPKC and loss of lateral Lgl induced by lack of Ral activity were suppressed by down-regulation of MSN/JNK signaling pathway. Conversely, the overexpression of msnWT in Ral70a FC did not enhance the Ral-induced aPKC (n=8/10 clones) and Lgl (n=6/6 clones) phenotypes (Figures S2C and D). These results suggest that during follicular epithelium remodeling Ral regulates both aPKC apical and Lgl lateral membrane localization by acting as an upstream negative regulator of MSN to JNK pathway (Figure 3G) and that the over activation of JNK signaling in the absence of Ral function results in loss of aPKC and Lgl membrane localization (Figures 2C and 2D).
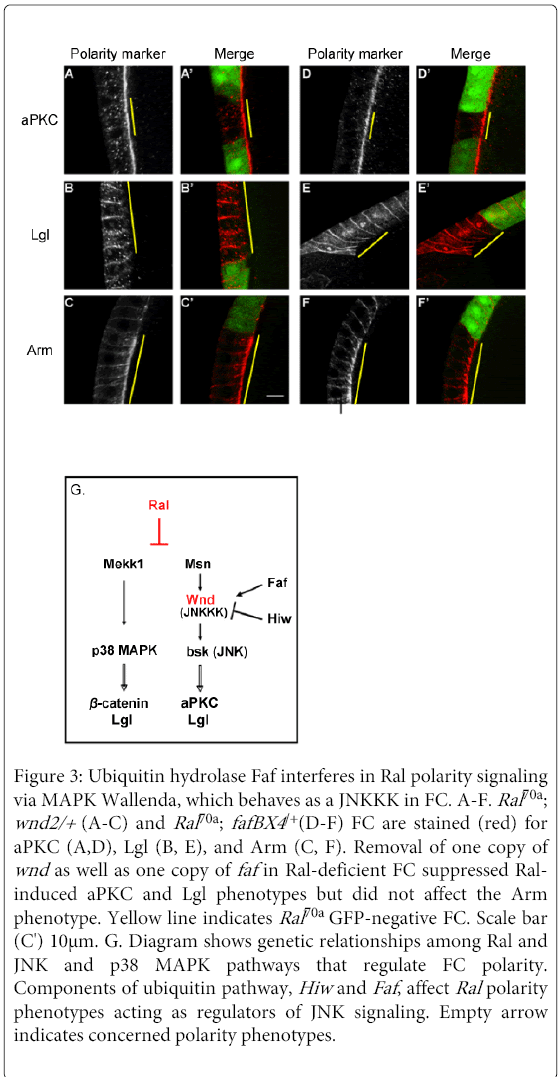
Figure 3: Ubiquitin hydrolase Faf interferes in Ral polarity signaling via MAPK Wallenda, which behaves as a JNKKK in FC. A-F. Ral70a ; wnd2/+ (A-C) and Ral70a ; fafBX4 /+(D-F) FC are stained (red) for aPKC (A,D), Lgl (B, E), and Arm (C, F). Removal of one copy of wnd as well as one copy of faf in Ral-deficient FC suppressed Ralinduced aPKC and Lgl phenotypes but did not affect the Arm phenotype. Yellow line indicates Ral70a GFP-negative FC. Scale bar (C') 10μm. G. Diagram shows genetic relationships among Ral and JNK and p38 MAPK pathways that regulate FC polarity. Components of ubiquitin pathway, Hiw and Faf , affect Ral polarity phenotypes acting as regulators of JNK signaling. Empty arrow indicates concerned polarity phenotypes.
To test whether over activation of JNK signaling induces polarity defects in follicle epithelium we overexpressed activated form of JNKK (hermipterous (hepCA)) under the control of Tj-Gal4 driver in all FC [30]. Polarization of FC as well as FC polarity organization during mitotic divisions was not affected, and both aPKC and Lgl were correctly localized from stage 2 till stage 8 egg chambers (data not shown). Hence, massive apoptosis was induced after st.8 and no egg chambers older than early-stage 9 was observed. Although these results confirmed that the JNK over activation doesn’t affect apical-basal polarity of FC during mitotic divisions the massive cell death of the post-mitotic follicular epithelium precluding any detailed analysis of its role during tissue remodeling.
Neither up- nor down-regulation of JNK pathway modified apical accumulation of Arm in Ral70a FC (Figures 2K and 2N and data not shown) suggesting that another Ral signaling pathway is implicated in Ral regulation of Arm localization. We therefore tested whether the p38 MAPK signaling modulates the Ral loss of function polarity phenotypes. The decrease of p38 MAPK signaling using mpk21 (encodes p38a MAPK) (n=13/16 clones) restored wild-type Arm localization at AJ in Ral-deficient FC (Figure 2H) and decreasing the protein level of p38 MAP3K (encoded by mekk1UR36) resulted in partial rescue of Ral-induced Arm phenotype (n=16/23 clones) (Figure S2I). Furthermore, decreasing of p38 MAPK signaling in Ral70aFC restored wild-type membrane localization of Lgl (n=16/17 for mpk21and n=14/16 clones for mekk1UR36) (Figures 2G and S2H). Ral-induced aPKC phenotype was not modified by down-regulation of p38 MAPK signaling (n=8/11 for mpk21/+, Figure 2F) and (n=11/12 clones for mekk1UR36/+, data not shown). The genetic interactions observed between Ral and p38 MAPK mutants indicate that Ral controls Arm and Lgl localizations by negatively regulating the p38 MAPK signaling pathway in FC during tissue remodeling (Figure 3G).
Thus, our genetic analysis reveals Ral function in aPKC apical localization by down-regulating JNK signaling and Ral function in Arm localization at AJ by down-regulating p38 MAPK signaling. Furthermore, Ral controls Lgl lateral localization by down-regulating both JNK and p38 MAPK pathways.
MAP kinase Wnd, ubiquitin hydrolase Faf and E3-ligase Hiw act as effectors of Ral activity in the regulation of epithelial polarity
Our analysis of Ral function in FC polarity organization enables us to further investigate the function of additional components of the JNK and p38 MAPK pathways. Wallenda (wnd) (an ortholog of vertebrate kinases DLK and LZK) is a MAP kinase kinasekinase (MAP3K) in JNK signaling (in Drosophila nervous system, [31] but also proposed to act as MAP3K in p38 MAPK signaling (in C.elegans, [32]. We analyzed whether a decrease of Wnd signaling affects Ral-dependent polarity phenotypes. The decrease of Wnd activity in Ral-deficient FC (Ral70a clones induced in wnd2/+ or in T155-Gal4>UAS-wndKD FC) suppressed Ral-dependent aPKC (n=17/26 and n=8/10 clones) and Lgl phenotypes (n=23/27 and n=7/7 clones) but did not modify Ral-dependent Arm phenotype (n=9/9 clones for wnd2) (Figures 3A-3C, S2J and S2K). These results indicate that Wnd acts as MAP3K in the JNK pathway in FC (Figure 3G).
A role for Wnd in JNK signaling was further confirmed by the analysis of two of its upstream regulators, High wire (hiw) and Fat facet (faf). Wnd has been shown to be down-regulated by the E3-ubiquitin ligase Hiw (mycBP2 in mammals) and up-regulated by the ubiquitin hydrolase Faf (USP9X in mammals) [31]. We observed that overexpression of Hiw or down-regulation of Faf (fafBX4/+context) in Ral70a FC restored wild-type localization of Lgl (n=14/15 or n=13/13 clones, respectively) and aPKC (n=11/11 or n= 20/27 clones, respectively) but did not affect Arm apical spreading and accumulation (n=13/17 clones for fafBX4) (Figures 3D-3F, S2L and S2M). Thus, we obtained the similar rescues as those produced by down-regulation of Wnd or of JNK signaling in Ral-deficient FC.
Ral loss of function suggests additional mechanisms for the maintenance of polarity in post-mitotic epithelial cells
Our analysis of Ral-deficient follicular epithelium demonstrates that Ral GTPase is required for the maintenance of AB polarity in polarized post-mitotic epithelial cells during morphogenetic reorganization of follicular epithelium. Lack of Ral signaling results in polarity defects: loss of cortical apical aPKC, and lateral Lgl, and Arm apical spreading and accumulation. Low fluorescent level of aPKC and increased of Arm observed in Ral mutant FC (Figure 1) indicates that lack of Ral function affect either the aPKC or Arm protein levels or their post-transcriptional modifications.
Ral supports polarity organization of FC by acting as an upstream negative regulator of MSN/JNK and MEKK1/p38 MAPK signaling pathways. Ral controls aPKC apical localization by down-regulating JNK signaling and Arm localization at AJs by down-regulating p38 MAPK signaling, whereas Ral regulation of Lgl membrane localization depends on both JNK and p38 MAPK pathways. While we cannot exclude that polarized secretion is affected in absence of Ral function, the rescue of Ral polarity phenotypes by JNK and MAPK loss of function suggests that over activation of JNK and/or p38 MAPK signaling in the absence of Ral function results in the defects of polarity organization in polarized post-mitotic epithelial cells. Moreover, our data demonstrated that in the absence of Ral signaling in polarized post-mitotic epithelial cells both basolateral anchoring and apical exclusion of Lgl can be maintained in the absence of apical aPKC, and apical mislocalization and accumulation of Arm takes place independent of Baz and DE-Cad wild-type localization in AJs.
In current models of polarity organization of epithelia, the regulation of Lgl by aPKC phosphorylation is critical in the control of the membrane domains integrity whereas Baz/Par3 determines the AJ domain by playing a key role in positioning the DE-Cad/β-catenin complex [12,15,33]. Baz/PAR-3 apical exclusion and its restriction to the apical side of the lateral domain are also regulated by the apical complex (Crumbs and aPKC) [15,17,34] and the phosphorylation of Baz on Serine 980 (pBazS780) by aPKC is essential for Baz apical exclusion and its localization at AJ [35,36].
Strikingly, our analysis reveals that in the absence of Ral function in polarized post-mitotic epithelial cells, aPKC becomes dispensable for maintaining Lgl basolateral localization. Indeed, in Ral70a clones both apical aPKC and lateral Lgl membrane localizations are compromised (Figure 1) and the decrease of p38 MAPK signaling in Ral70a FC can restore Lgl localization at the basolateral domain in the absence of apical aPKC (Figure 2). Thus, in the absence of Ral signaling both Lgl cytoplasmic release and Lgl basolateral membrane anchoring seem independent from aPKC apical localization and aPKC kinase activity. These results indicate the existence of additional mechanisms of Lgl lateral localizationin the absence of aPKC polarization that can be achieved by the polarized basolateral delivery of Lgl or by the apical exclusion of Lgl by a yet unknown kinase.
In addition, in the absence of Ral signaling the wild type localization of Baz at AJ was observed whereas apical aPKC membrane localizations was compromised in Ral70a FC (Figure S1A) as well as in Ral70a;mpk2mut/+ FC (data not shown) indicating that Baz distribution is not controlled by apical aPKC and the phosphorylation of Baz by aPKC is not essential for Baz localization during late oogenesis.
Furthermore, in the absence of Ral activity in polarized post-mitotic FC Arm distribution becomes largely independent of DE-Cad and Baz. In fact, Arm apical spreading and accumulation is observed in spite of the fact that both Baz and DE-Cad are correctly localized at AJ (Figure S1). Thus, the interactions DE-Cad/Arm and Baz/Arm seem to be not sufficient to properly localize Arm suggesting that in polarized epithelial cells Arm might be recruited to the cell membrane independently of DE-Cad localization.
Accordingly, analysis of AJ organization and dynamics in polarizing epithelial cells and in polarized epithelial cells demonstrated that in the polarized epithelial cells during tissue remodeling Baz localization was largely independent of aPKC [17] and Arm localization at AJs are less controlled by Baz [34] as well as Arm membrane dynamics could be distinct from the one of DE-Cad [37,38].
Thus, analysis of Ral loss of function during epithelial morphogenesis in Drosophila ovaries reveals that additional mechanisms might control Lgl and Arm distribution in polarized epithelial cells during tissue remodeling.
Our data demonstrate that Ral GTPase is required for the maintenance of AB polarity during tissue remodeling associated with cell shape changes. Lack of Ral activity during morphogenetic reorganization of follicular epithelium causes loss of apical aPKC, loss of lateral Lgl and its cytoplasmic mislocalization, and Arm/β-catenin apical spreading and accumulation. Our analysis demonstrates that Ral regulates polarity organization of FC acting as an upstream negative regulator of MSN/JNK and MEKK1/p38 MAPK signaling pathways. Ral controls aPKC apical localization by down-regulating JNK signaling and Arm/β-catenin localization at adherens junctions by down-regulating p38 MAPK signaling, whereas Lgl membrane localization depends on down-regulation of both JNK and p38 MAPK signaling pathways. Furthermore, analysis of Ral loss of function during epithelial morphogenesis in Drosophila oogenesis demonstrates that additional mechanisms control Lgl and Arm distribution in polarized epithelial cells during tissue remodeling.
We thank Janice Fischer, Aaron Di Antonio, Yong Rao, Hideki Inoue, Takashi Adachi-Yamada as well as VDRC, Bloomington and NIG-Fly Stock Centers for providing flies; Antoine Guichet, Manzoor Bhat, Andreas Wodarz, Juergen Knoblich for providing primary antibodies, and Jean-René Huynh for his advice and discussion. This work was supported by grants from ANR-0338-02, ANR -0290-01, INCA -PLBIO-03-IC-1; by the FRM (26329) and the INCA (4731);by the NWO-Rubicon grant to F.B., by grants to Y.B. from the ARC (4830), the ANR (BLAN07-3-207540, Morpho Dro) and the ERC Starting Grant (CePoDro 209718); by the CNRS, INSERM and the Curie Institute. C.B. was further supported by fellowships from MRT and ARC