Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2017) Volume 6, Issue 3
Noscapine (narcotine) is an opium alkaloid, isolated from Papaver somniferum L. Noscapine is being used in clinical trials for curing cancer. Till date researchers mainly discussed the erythro form of noscapine but in the present work the authors have discussed the erythro as well as threo form of noscapine. In the present work, library of 11 derivatives of noscapine from each of threo- and erythro- isomer was created and virtually screened for their chemotherapeutic efficacy on Tyrosine Kinase Domain from Epidermal Growth Factor Receptor (PDB-1M14). The interaction between Tyrosine Kinase Domain of Epidermal Growth Factor Receptor and noscapines was studied by means of docking based on lowest binding energy and top pharma score and Molecular Dynamic (MD) simulation. It was found erythro form aminonoscapine (13) have higher binding affinity against the Tyrosine Kinase Domain and further, MD simulation was performed. Compound 13 showed acceptable hydrophobicity and solubility value. Further, threo form of nitronoscapine (11) also showed good interaction based on docking, simulation and comes out to be acceptable like a drug based on hydrophobicity and solubility value.
<Keywords: Epidermal growth factor receptor; Threo and erythro of noscapine; Docking; Simulation
Noscapine is a heterocyclic compound and isolated from the poppy plant [1]. Noscapine is found to be a major alkaloid in the latex of opium poppy and it is being used as cough suppressant from last many decades and it attracted researchers for the investigation as a potential anticancer drug [2-4]. Noscapine is used to stop cellular proliferation and induces apoptosis or the process cell death in nonsmall cell lung cancer cells either alone or in combination with other chemotherapeutic drugs. Currently, it is used in Phase I/II clinical trials for cancer chemotherapy [5,6].
The epidermal growth factor receptor is a member of the ErbB family. It is a subfamily of four closely related receptor tyrosine kinases: EGFR (ErbB-1), HER2/neu (ErbB-2), Her 3 (ErbB-3) and Her 4 (ErbB- 4) [7]. The loss of control over these vital cellular processes is a hallmark of oncogenesis [8]. For instance, aberrant signaling from overexpressed growth factor receptor ErbB2 is causal in approximately 30% of invasive breast cancers [9-14]. Binding of extracellular conpounds to the ligandbinding domain of the receptor leads to formation of active homo- or hetereodimers, inducing C-terminal autophosphorylation of EGFR and activation of docking proteins, such as GRB2 or SHC1 [15-17]. The most frequent EGFR mutations in lung adenocarcinoma are located in the kinase domain of the receptor and lead to ligand-independent, constitutive kinase activation [16-21]. Better water solubility and feasibility for oral administration of noscapine over many other anticancerous drugs [22]. Hence, among the various anticancerous agents that perturb protein dynamics, noscapines constitute an emerging class of compounds receiving considerable attention of researchers [23,24].
In the present work, authors have designed several analogues of noscapine (each of erythro and threo) better therapeutic indices. A comparative study with other reported ligand molecules was evaluated for their chemotherapeutic efficacy on Tyrosine Kinase Domain from Epidermal Growth Factor Receptor.
The work has been divided into three parts i.e. molecular docking, protein dynamic simulation and solubility analysis.
Docking preparation
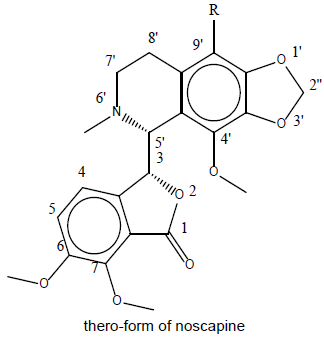
Designing of compound: Noscapine exist in two isomeric form viz., erythro and threo form. Several derivatives of noscapine were prepared by substituting at the 9’ position mentioned in Table 1.
| Parent molecule | S.N. | R= | Parent molecule | S.N. | R= |
|---|---|---|---|---|---|
 |
1 | -H | 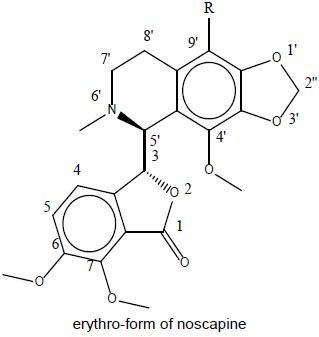 |
12 | -H |
| 2 | -NH2 | 13 | -NH2 | ||
| 3 | -Br | 14 | -Br | ||
| 4 | -CHO | 15 | -CHO | ||
| 5 | -COCl | 16 | -COCl | ||
| 6 | -COOH | 17 | -COOH | ||
| 7 | -Cl | 18 | -Cl | ||
| 8 | -CH2Cl | 19 | -CH2Cl | ||
| 9 | -F | 20 | -F | ||
| 10 | -CH2OH | 21 | -CH2OH | ||
| 11 | -NO2 | 22 | -NO2 |
Table 1: Library of noscapines (threo and erythro) A library of reported potential compound (23-33) was created as in Table 2. They have been taken from the protein databank (www.rcsb.org).
A library of reported potential compound (23-33) was created as in Table 2. They have been taken from the protein databank (www. rcsb.org).
| S.N. | Compound Name | S.N. | Compound Name | S.N. | Compound Name | S.N. | Compound Name |
|---|---|---|---|---|---|---|---|
| 23 | [6,7-Bis(2-methoxy-ethoxy)quinazoline-4-yl]- (3-ethynylphenyl)amine |
24 | 1,2,3,4-Tetrahydrogen Staurosporine |
25 | 2-(Acetylamino)-2-deoxy--D-glucopyranose |
26 | 6-{4-[(4-ethylpiperazin-1-yl)methyl]phenyl}- n-[(1R)-1-phenylethyl]-7h-pyrrolo[2,3-d]pyrimidin- 4-amine |
| 27 | 6-deoxy-beta-L-galactose |
28 | -D-Nannose |
29. | β-D-Mannose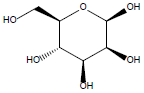 |
30 | Gefitinib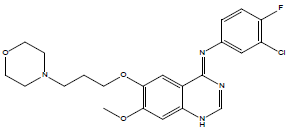 |
| 31 | N-[4-(3-bromo-phenylamino)-quinazolin-6-yl]-acrylamide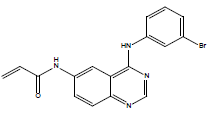 |
32 | N-acetyl glucosamine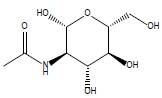 |
33 | Thiophosphoric acid-O-((adenosyl-phospho)phospho)- S-acetamidyl-diester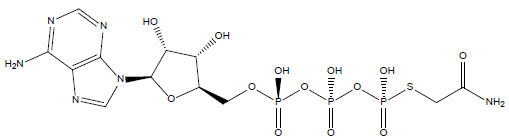 |
||
Table 2: List of previously docked compound with the same protein.
3D Sketching and geometry optimization: All thirty three compounds were prepared with CS Chemdraw and saved in appropriate format. Geometry of the all molecule were optimized with the help of Chem 3D by applying the molecular mechanics (MM2) as a force field via performing the minimization of energy, where the RMS Gradient is set to 0.010. The optimized compounds were further saved in suitable format, which is used in docking.
Protein preparation: Protein preparation plays crucial role in docking. The preparation of protein was done using Molegro Molecular Viewer (MMV 2.5). Different aspect were studied like assigning of bonds, missing bond order and hybridization, missing explicit hydrogen, missing charges, flexible torsion in compounds, tripos type atoms. Finally, the prepared protein is used for molecular docking.
Molecular docking
All threo and erythro derivatives of noscapines as in Table 1 and reported molecule as in Table 2 were docked into the binding pocket of the tyrosine kinase domain of EGFR protein (PDB ID - 1M14) using iGEMDOCK (Generic Evolutionary Method for molecular Docking) software [25]. This computational tool has a program for computing the conformation of the small molecules and also its orientation towards the active site of the tyrosine kinase domain of EGFR. The binding pockets of the PDB-1M14 was defined to include the amino acid residues in the region of 8˚A radius on the binding site of tyrosine kinase domain of EGFR.
In the present work, the set parameters of this computational tools are, population size (n=200), generations (g=70) and number of solutions for each compound (s=2). Interaction data was generated and shorted by applying the default parameters of energy and z-score. Top compound was selected on the basis of energy and z-score so that lead out will be most potent one.
EBinding = vdW + Hbond + Elec
vdW stands for van der Waal energy, Hbond stands for hydrogen bonding energy and Elec stands for electro statistic energy.
The z-score value is a parameter to measure the interaction between the protein and noscapine on pharmacological basis. The pharmacological scoring function is given as;
Epharma (z-score) = EGEMDOCK + E(E)pharma + 2E(H)pharma + 0.5E(V)pharma
Where EGEMDOCK is the docked energy of GEMDOCK and E(E)pharma, E(H)pharma, and E(V)pharma are the pharmacological scores of electrostatics, hydrogen-bonding, and vdW interactions, respectively.
The modelling of docked poses was done by MMV 2.5 and we also uses the LIGPLOT+ v-1.4.5, to confirm the same and analyse the protein compound interaction more clearly and also to confirm the H-bonding length of interaction between amino acid residues of protein and compound.
MD Simulation
The nanoscale simulation method can reveal the fundamental material structure and response characteristics to predict the macroscopic response, behaviour of protein and protein compound complex. In this study, GROMACS 5.1.4 was used to simulate the protein [25] and LIGRO was used to simulate protein-compound complex [26]. The force-field parameters used is OPLS-AA (Optimized Potentials for Liquid Simulations-All Atom). LIGRO was developed by Luciano Porto Kagami et al., [26], it is a combination of Amber, [27] ACPYPE [28] and PLIP [29]. Along with GROMACS. The SPC (simple point charge) water model was used for simulation with the cubic box type. The system was neutralized by adding the counter ion (Na+ and Cl-) and concentration was set to 0.15 M. The steepest decent algorithm with 1000 steps has been used for the minimization of energy to release the conflict contacts.
The MD simulation studies comprised of two phases, (i) ensemble equilibration and (ii) MD production. To control the temperature, NVT ensemble is used with leap-frog integrator and temp was set to 300K and to control the pressure the NPT ensemble is used with leapfrog integrator and pressure was set to 1bar and number of steps (ns) is set to 500000 (1000 ps) for both. Now for the second phase, the MD simulation were carried out with the ns equals to 500000 (1000 ps) with leap-frog integrator and co-ordinates were recorded at every 10 ps for the analysis.
Log P and Log S calculation
Lipophilicity is an indicator to understand physicochemical property of a potential drug. It plays a role in solubility, absorption, membrane penetration, plasma protein binding, distribution, central nervous system penetration and partitioning into other tissues or organs such as the liver and has an impact on the routes of clearance. Log P is used as a measure of lipophilicity and it is the partition coefficient of a molecule between an aqueous and lipophilic phases, usually octanol and water.
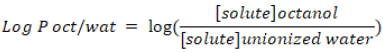
Solubility is another common physicochemical parameter for drug development. Aqueous solubility of the molecule reflects the bioavailability. The poor solubility of molecules limits the absorption of compound from the gastrointestinal tract. Web server (www.vcclab. org) was used to calculate the Log S and Log P values of all compounds (1-33) are listed in the Tables 3-5.
| Compound | Total Energy | VDW | HBond | Elec |
|---|---|---|---|---|
| 13 | -102.771 | -87.3142 | -15.4566 | 0 |
| 21 | -94.0027 | -85.6051 | -8.39766 | 0 |
| 12 | -93.9668 | -83.4948 | -10.472 | 0 |
| 11 | -93.259 | -70.5126 | -22.2678 | -0.47868 |
| 22 | -92.7138 | -76.23 | -16.4838 | 0 |
| 14 | -91.9388 | -66.6272 | -25.3116 | 0 |
| 15 | -90.5276 | -71.8545 | -18.6732 | 0 |
| 16 | -89.6727 | -62.8 | -26.8726 | 0 |
| 10 | -89.4758 | -72.877 | -16.5988 | 0 |
| 20 | -88.6383 | -70.737 | -17.9013 | 0 |
| 5 | -88.1461 | -82.5475 | -5.59855 | 0 |
| 17 | -87.3615 | -66.4635 | -20.898 | 0 |
| 19 | -86.6471 | -75.7417 | -10.9054 | 0 |
| 2 | -85.7716 | -63.0076 | -22.764 | 0 |
| 4 | -84.9927 | -72.3875 | -12.6051 | 0 |
| 18 | -84.7043 | -65.7218 | -18.9825 | 0 |
| 8 | -83.4453 | -64.0964 | -19.3489 | 0 |
| 6 | -83.0829 | -81.3006 | -1.78232 | 0 |
| 7 | -82.6244 | -64.4359 | -18.1884 | 0 |
| 1 | -82.3567 | -67.4153 | -14.9414 | 0 |
| 9 | -80.6864 | -78.8994 | -1.78697 | 0 |
| 3 | -80.2376 | -61.7299 | -18.5077 | 0 |
Table 3: Interaction of noscapines with tyrosine kinase domain of EGFR having PDB ID-1M14A.
| Compound | Total Energy | VDW | HBond | Elec |
|---|---|---|---|---|
| 33 | -116.51 | -64.5678 | -45.9509 | -5.99083 |
| 26 | -115.028 | -97.0767 | -17.9517 | 0 |
| 31 | -112.749 | -82.5469 | -30.2019 | 0 |
| 30 | -103.053 | -90.5867 | -12.466 | 0 |
| 23 | -102.365 | -95.5622 | -6.8025 | 0 |
| 24 | -97.1494 | -77.7756 | -19.3738 | 0 |
| 32 | -81.0784 | -53.5265 | -27.5519 | 0 |
| 25 | -74.0972 | -50.594 | -23.5032 | 0 |
| 29 | -73.4745 | -39.4164 | -34.0581 | 0 |
| 28 | -71.3851 | -40.8233 | -30.5618 | 0 |
| 27 | -68.4251 | -40.4251 | -28 | 0 |
Table 4: Interaction of eleven compounds (compound number 23-33) with tyrosine kinase domain of EGFR having PDB ID-1M14.
| C.N. | Log P | Log S | C.N. | Log P | Log S | C.N. | Log P | Log S |
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | -3.36 | 12 | 2 | -3.36 | 23 | 3.13 | -4.64 |
| 2 | 1.57 | -2.75 | 13 | 1.57 | -2.75 | 24 | 3.17 | -4.17 |
| 3 | 2.85 | -4.05 | 14 | 2.85 | -4.05 | 25 | -2.06 | 0.08 |
| 4 | 1.6 | -3.28 | 15 | 1.6 | -3.28 | 26 | 4.38 | -4.43 |
| 5 | 2.39 | -3.13 | 16 | 2.19 | -4.09 | 27 | -2.39 | 0.7 |
| 6 | 1.61 | -3.03 | 17 | 1.61 | -3.03 | 28 | -2.57 | 0.64 |
| 7 | 2.76 | -4.02 | 18 | 2.76 | -4.02 | 29 | -2.57 | 0.64 |
| 8 | 2.84 | -4.11 | 19 | 2.84 | -4.11 | 30 | 3.42 | -4.91 |
| 9 | 2.19 | -3.49 | 20 | 2.19 | -3.49 | 31 | 3.49 | -4.66 |
| 10 | 1.39 | -2.92 | 21 | 1.39 | -2.92 | 32 | -2.6 | 0.06 |
| 11 | 2.06 | -3.35 | 22 | 2.06 | -3.35 | 33 | -0.87 | -2.15 |
Table 5: Log P and Log S values of all compounds as in Table 1 & 2 (1-33).
The whole result and discussion is divided into three major part i.e., docking, dynamic simulation and solubility analysis.
Molecular docking
Molecular docking is considered as a popular method. It is used for structure-based virtual screening for finding the potent compounds towards a particular target protein. iGEMDOCK is a computational tool and uses an empirical scoring function as well an evolutionary approach [30-34]. The results obtained were based on the total energy and is summation of energy contributed due to electrostatic, steric, and hydrogen-bonding. Steric and hydrogen bonding uses a linear model. It recognizes potential complexes rapidly by means of scoring function and change in energy of docking. The idea behind this evolutionary approach is to design plenty of operators that cooperate using a family competition paradigm that is similar to a local search procedure. Herein, a theoretical model based on atomic level interactions was designed to study the potential noscapine against tyrosine kinase domain of EGFR.
Result of docking has been considered, if the root-mean-square derivation (RMSD) between the docked pose of compound and X-ray crystal structures of protein was found to be ≤ 2 Å. Although, the of the interaction of compounds (1-22) with tyrosine kinase domain of EGFR was studied at a distance of 4 Å and the result in Table 3, where the highest negative value of binding energy has been taken under consideration. Further, reported molecules significantly interacted with tyrosine kinase domain of EGFR (PDB-1M14) seen from result obtained as in Table 4. Only compound 33, 26, 31, and 30 showed better binding than compound 13.
In molecular docking, the amino acid showed effective binding with compound 13. This new binding domain may serve as valuable pocket in tyrosine kinase domain of EGFR. The binding domain of tyrosine kinase domain of EGFR for compound 13 is composed of LEU-679, VAL-745, ARG-807, PRO-675, ASN-676, GLN-677, LEU- 679, ALA-743, SER-744, VAL-745, ARG-807, and for compound 11 is composed of LEU-679, ASP-746, ARG-752, GLN-677, ALA-678, LEU- 679, ALA-743, VAL-745, ASP-740, ARG-752 as shown in Graph 1a and 1b respectively. These binding domains are acquired by noscapines 13 and 11. They showed highest negative binding energy among the all derivatives of erythro and threo having value -102.771 and -93.259 respectively. The pharma score or Z-score value for these compound are also found highest for the noscapine derivatives, the docked poses are given in Figure 1a-f.

Graph 1: (a and b) Total binding energy (δGbind) contribution of amino acids within and around binding site (8 Å of the docked compound) of EGFR involved in interaction with noscapine derivatives no. 13 and 11 respectively. Ala 678 for compound 13 and Asp 746 for compound 11 is a consistently important contributor to the binding energy.

Figure 1: (a and b) Docked pose of compound no. 13 and 11 with tyrosine kinase domain of EGFR (PDB-1M14) in 3D view showing the cavity within the protein and binding site respectively; (c and d) Docked pose of compound no. 13 and 11 with tyrosine kinase domain of EGFR (PDB-1M14) in 2D view showing the interaction of compound with selected amino acid residues of protein with the number of possible H-bonding interaction respectively; and (e and f) LIGPLOT of compound no. 13 and 11 with PDB-1M14 in 2D view clearly showing the H-bonding interaction length, 3.03 and 2.60 Å respectively and the interacting site of compound 13 and 11 is different.
Molecular dynamic simulation
Molecular dynamics (MD) is widely used and effective way in fields of molecular modeling. MD simulation represents the flexibility of both the compound and protein more effectively. The folding patterns of 1M14 were analysed in water. The stability of the 1M14 and 1M14- compound complex were assessed on the basis of calculated radius of gyration (Rg), Root Mean Square Deviations (RMSD), Root Mean Square Fluctuations (RMSF) and Mean Square Deviation of protein (MSD) during the course of 1000 ps. For the these systems, significant similarities were found in the Rg, RMSD, RMSF and MSD values in some region of protein and protein-compound complex as shown in Figure 2. The higher disruptions occur in case of compound no. 11 as compare to compound no. 13.

Figure 2: (a-c) Time dependence of Rg of protein, protein-13 and protein-11; (d-f) Time dependence of RMSD of protein, protein-13 and protein-11; (g-i) RMSF values of protein, protein-13 and protein-11; (j-l) Time dependence of MSD of protein, protein-13 and protein-11.
As we know that the radius of gyration of protein is a measure of its compactness. If a protein is well folded, then it will likely maintain a relatively steady value of Rg. If protein were unfold, its Rg will change over the time. In Figure 2a-c, the Rg value of protein and proteincompound (13 or 11) complex were ploted from 0 to 1000 ps. It was found that the four curve for protein and protein compound complex at the value of about 1.42, 1.66, 1.76 and 1.99. In case of protein, these all four curve remained in invariant position while, In case of protein complexed with compound 13 the fluctuation remains small in the three curve but for the fourth curve a slight fluctuation was found. In case of protein complexed with compound 11, we found that significant change in the timescale of the Rg values was found. The above result clearily indicate that there will be more stabilization in case of protein complexed with the compound 13.
Root mean square deviation is the most commonly used quantitative measure of the similarity between two superimposed atomic coordinates. RMSD can be calculated for any type and subset of atoms. In Figure 2d-f, the RMSD values of protein and proteincompound (13 or 11) complex were plotted from 0 to 1000 ps for the atoms of protein and protein-compound complex. They found that the RMSD value for protein is remains invariant around 0.18 nm and for the protein complexed with compound 13 is also remains invariant about 0.18 nm, while in case of protein complexed with compound 11, some fluctuation has been found. The above result also confirmed the more stability of protein complexed with compound 13.
When a dynamical system fluctuates about some well-defined average position, the RMSD from the average over time can be referred to as the root mean square fluctuation (RMSF). The size of this fluctuation can be measured using the computational tools via MD simulation. Herein, the RMSF study of around 350 amino acid residues were performed from the residue 650 to 1000 (approx.) of the whole protein molecule, these residues also belongs to the binding pocket of protein. (Figure 2g) More fluctuation means more interaction of compound (13 or 11) with the protein. (Figure 2h and 2i) The fluctuation in the region of 650 to 1000 amino acid residue containing LEU-679, VAL-745, ARG-807, ALA-674, PRO-675, ASN-676, GLN- 677, ALA-678, LEU-679, TYR-740, ALA-743, SER-744, VAL-745, ASP-746, LEU-753, ARG-807 and LEU-837 support the binding interaction of docking experiment. Herein, more fluctuation in the case of compound 11 as compare to compound 13 was observed.
Mean square deviation also gave the information about the structure stability of the protein molecules. Less the deviation more will be stability of protein molecule. In Figure 2d-f, the plots MSD of protein and protein-compound (13 or 11) complex were also plotted for the 0 to 1000 ps. It can be clarified that the more stabilization of protein complexed with compound 13 as compare to the compound 11.
Hydrophobicity and solubility
Lipophilicity indicates the permeability of the compounds through the various biological membranes and the value of octanol-water partition coefficient log P should not greater than 3 and not less than -0.4. The Log P value of compounds 33, 26, 31, and 30 are -0.87, 4.38, 3.49 and 3.42 respectively. These values suggested that these molecules can’t be used as drug molecule, whereas the log P values of compound 13 and 11 are under the acceptable range which is 1.57 and 2.06. The aqueous solubility of a compound significantly affects its absorption and distribution characteristics. Typically, a low solubility goes along with a bad absorption and therefore the general aim is to avoid poorly soluble compounds for the drug. Drugs with log S value less than 0 and greater than -4 is absorbed in blood and tissue. It has been found that compound 13 and 11 have the acceptable log S value which is -2.75 and -3.35 respectively.
The interaction between tyrosine kinase domain of EGFR and noscapines was studied computationally. Stabilization of tyrosine kinase domain of EGFR by noscapines achieved successfully via molecular docking by performing drug screening mode. It was found that noscapines showed significant interactions with the protein predominantly via their hydrogen bonding, van der Waals interactions and electrostatic. The favourable electrostatic and van der Waals interaction energies calculated for the association between tyrosine kinase domain of EGFR and noscapine showed remarkable point to accept this model as a novel work. Compound 13, erythro form of aminonoscapine showed maximum binding among the deigned (1- 22) compounds. The MD simulation results clearly verify the effective docking of noscapine. The result gave an indication that the noscapine could be used to prevent dimerization of protein.