Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2013) Volume 2, Issue 2
In recent years, a promising new approach uses the anti-angiogenesis properties of certain molecules to try to block cancer by depriving tumors of nutrients and oxygen they need to grow. Endothelial cells, specialized in the development of new blood vessels, are the target of most anti-angiogenic strategies, Or methionine aminopeptidase (MetAp) type 2 is a member of a family of proteins that regulates the growth of these endothelial cells. The molecular docking program, GOLD, was developed to assist in the development of molecules with therapeutic activity. It has been used to study the inhibition of 1QZY, human methionine aminopeptidase type 2, by bengamide derivatives, with the aim of discovering new anti-angiogenic drugs. The evaluation of the affinity of these molecules brought to light those presenting the best inhibitive effect. It is about the compound 16, the value of the score of which is 135.35.
<Keywords: Tumor; Anti-angiogenesis; Methionine aminopeptidase; Docking; GOLD; RMSD
Molecular docking techniques have shown great promise as a new tool in the discovery of novel small molecule drugs for targeting proteins, because of their speed, economy and increasing reliability [1].
In this context, methionine aminopeptidase (MetAp) is a dimetalloprotease that catalyzes the removal of N-terminal methionine in newly formed polypeptide chains. MetAps are found in both eukaryotic and prokaryotic cells, and two types of MetAps are recognized from amino acid sequence comparisons and structural studies: type 1 (MetAp-1) and type 2 (MetAp-2) [2].
The human MetAp2 isoform has been of considerable interest as a target for cancer chemotherapy, after the discovery that the antiangiogenic natural compound, fumagillin, is a covalent inhibitor selective for MetAP-2. On the basis of the antiangiogenic activity observed in vitro, a fumagillin analog TNP-470 has progressed to clinical trials; however, the compound has demonstrated dose limiting neurotoxicity. A second family of natural products, the bengamides, has recently been identified as inhibitors of both MetAp isoforms [3].
The purpose of this work is to test the reliability of the molecular docking program GOLD (Genetic Optimization for Ligand Docking), used in this study by determining the RMSD (root mean square deviation), then use this program to search for a new inhibitor of methionine aminopeptidase through similar of bengamide [4], with a degree of similarity up to 95% from the data bank PubChem.
Molecular docking
Docking is one of the commonly used computational methods in structure based drug design. Docking is the process of fitting of the ligand into the receptor. It not only gives an idea about how the ligand is going to bind with the receptors, but also about up to what extent conformational changes can be brought in the receptor structure. Docking comprises two distinct tasks, the first being the prediction of favorable binding geometries for a small molecule in the binding site of a target protein, and secondly, the estimation of the binding free energy of the complex so formed, also referred to as scoring.
Docking accuracy reflects an algorithm’s ability to discover a conformation (pose) and alignment of a ligand relative to a cognate protein that is close to that experimentally observed, and to recognize the pose as correct. Scoring accuracy is the ability to correctly predict the rank order of binding affinities of ligands to a particular protein [5-7].
GOLD v 5.0.1 (2011)
GOLD [8] is one of the most successful docking program and widely used. It is based on three major parts:
A scoring function to rank different binding modes: The Goldscore function is a molecular mechanics–like function with four terms:
GOLD Fitness=Subtext+S vdw_ext+S hb_int+S vdw_int
Where Subtext is the protein–ligand hydrogen-bond score and Svdw_ext is the protein-ligand van der Waals score. Shb_int is the contribution to the Fitness due to intramolecular hydrogen bonds in the ligand; Svdw_int is the contribution due to intramolecular strain in the ligand.
A mechanism for placing the ligand in the binding site: GOLD uses a unique method to do this, which is based on fitting points; it adds fitting points to hydrogen bonding groups on protein and ligand, and maps acceptor points on the ligand on donor points in the protein, and vice versa. Additionally, GOLD generates hydrophobic fitting points in the protein cavity onto which ligand CH groups are mapped.
3. A search algorithm to explore possible binding modes; GOLD uses a genetic algorithm (GA).
Structure of the enzyme
Structure of the enzyme derived from the PDB "Protein Data Bank". The PDB (Protein Data Bank) is the single worldwide archive of structural data of biological macromolecules, established in Brookhaven National Laboratories (BNL) in 1971 [9]. It contains structural information of the macromolecules determined by X-ray crystallographic, NMR methods etc.
The 1QZY structure was chosen for this study, because it represents a compromise between good resolution (1.60), and the presence of an inhibitor (TDE).
Lipinski rule
Each drug must comply with several basic criteria, such as low cost of production, be soluble, stable, but must also conform to the schedules associated with its pharmacological properties of absorption, distribution, metabolism, excretion and toxicity (ADME/Tox) [10], which is based on the rule of five made by Christopher Lipinski [11]. The “rule of 5” states that: poor absorption or permeation is more likely when:
- There are more than 5 H-bond donors
- The MWT is over 500
- The Log P is over 5
- There are more than 10 H-bond acceptors
In addition, Veber has introduced two supplementary criteria to what is now commonly called "the rule of five". Surface area of the polar compound must be less than 140 Å and the number of rotatable bonds must be less than 15 [12].
The ability of molecular docking software GOLD
Program performance has been evaluated on 150 complexes available in the PDB. The docked binding mode is compared with the experimental binding mode, and a root-mean-square distance (RMSD) between the two is calculated (by GOLD); a prediction of a binding mode is considered successful if the RMSD is below a certain value (usually 2.0 Å) [5,8,13]. In the graph, the results are given in percent.
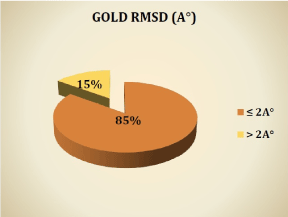
Graph: Results in % of docking by GOLD.
We notice from these results that the program GOLD reproduces well the experimental data. Indeed, 85% of RMSD values are less than 2 Å. It is clear from this graph that the RMSD values are consistent with the results of Abdelouahab C and Abderrahmane B [5], Vieth et al. [13] and Gabb et al. [14], showing that any program the docking is successful when the RMSD is less than 2 Å. This is also consistent with the results obtained by Zaheer-ul-Haq et al. [15], where six docking programs were used: FRED, GOLD, MOE, AutoDock, FlexX and Surflex-Dock, for a comparative study to determine their ability to reproduce poses via the experimental RMSD. FRED was the best followed by Surflex-Dock and GOLD. In the same year [16] evaluated the performance of four programs: GOLD, AutoDock, Surflex-Dock and FRED by calculating the RMSD, the best results were obtained by GOLD and FRED. In addition, our results confirm the results obtained by Hioual et al. [17], where the program GOLD has been tested with the software FlexX; GOLD showed a large performance to reproduce the experimental tests with 64.05% of the RMSD values less than 2 Å. Thus, this software can be used to predict the interactions MetAP-inhibitors.
Docking bengamide derivatives
The docking by GOLD of the bengamide and its similar gave the following results, which are presented in the Table 1.
| N | Inhibitors | Fitness (score) |
|---|---|---|
| 1 | BENGAMIDE | 115.28 |
| 2 | CID_2322 | 109.94 |
| 3 | CID_2323 | 115.06 |
| 4 | CID_448235 | 112.71 |
| 5 | CID_5353435 | 101.13 |
| 6 | CID_638810 | 85.00 |
| 7 | CID_6444155 | 113.15 |
| 8 | CID_6476094 | 114.28 |
| 9 | CID_6476095 | 114.02 |
| 10 | CID_9975670 | 107.59 |
| 11 | CID_10090681 | 99.27 |
| 12 | CID_10316350 | 98.63 |
| 13 | CID_11057452 | 96.50 |
| 14 | CID_11245731 | 115.40 |
| 15 | CID_11246226 | 116.79 |
| 16 | CID_11249087 | 135.35 |
| 17 | CID_11383607 | 114.47 |
| 18 | CID_11639347 | 113.30 |
| 19 | CID_11682243 | 103.99 |
| 20 | CID_21637220 | 110.54 |
| 21 | CID_24782418 | 118.73 |
| 22 | CID_24782420 | 113.17 |
| 23 | CID_24840580 | 94.27 |
| 24 | CID_24840583 | 99.47 |
| 25 | CID_44124768 | 110.89 |
| 26 | CID_44124877 | 117.55 |
| 27 | CID_44124881 | 111.65 |
| 28 | CID_44125009 | 119.34 |
| 29 | CID_44125012 | 114.49 |
| 30 | CID_44125146 | 106.05 |
| 31 | CID_44125148 | 105.36 |
| 32 | CID_44125149 | 111.51 |
| 33 | CID_44347653 | 108.49 |
| 34 | CID_44347702 | 115.79 |
| 35 | CID_44347715 | 109.43 |
| 36 | CID_45027798 | 108.15 |
| 37 | CID_46915399 | 115.60 |
| 38 | CID_46915400 | 118.76 |
| 39 | CID_46915538 | 109.65 |
| 40 | CID_46915539 | 114.04 |
| 41 | CID_46915540 | 97.08 |
| 42 | CID_46936660 | 95.39 |
| 43 | CID_53260818 | 99.07 |
Table 1: Results of the docking bengamide and its similar 95%.
Of the 43 inhibitors tested the similar N° 16 (Table 1) forms the most stable complex protein-ligand, so it has the best inhibitory effect with fitness value of 135.35.
Lipinski rule
Before starting the study of interactions between the enzyme MetAP and the compound 16, it is necessary to evaluate the parameters for validation as an antibiotic (Table 2). These indices were calculated under the code "Molinspiration." It used to draw molecules and calculate molecular properties important (log P, polar surface, the number of donors and acceptors the hydrogen bond, ..., etc.) directly on a web page.
| Compound | Molecular weight(g/mol) | H Donors | H Acceptors | Log P | Flexible bonds |
|---|---|---|---|---|---|
| 16 | 501.65658 | 5 | 8 | 0.8 | 15 |
Table 2: Lipinski rule.
The results in Table 2 show that the molecule 16 used in this study responds to the rule of Lipinski.
Interaction MetAP-similar 16
To study the mode of interaction of different inhibitors with the active site of the human methionine aminopeptidase (MetAp- 2) by molecular docking, we used the latest version of the program GOLD which can show the Van Der Waals contacts and the hydrogen bonds that are the most important among the weak links.
The Figure 1 shows that the inhibitor 16 penetrates well into the active site of the enzyme, forming 4 hydrogen bonds represented in the Table 3.
| N | Residues involved | Atom of amino acid | Ligand atom | Distance Å |
|---|---|---|---|---|
| 1 | Asp251 | OD2 | O5 | 2.949 |
| 1 | Asp262 | OD2 | O7 | 2.413 |
| 2 | Glu364 | OE1 | N2 | 3.006 |
| O4 | 2.851 |
Table 3: Hydrogen bonds.
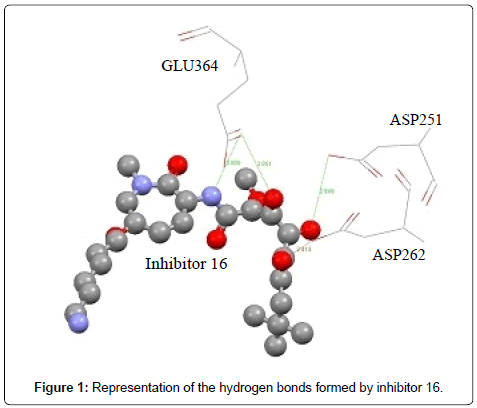
Figure 1: Representation of the hydrogen bonds formed by inhibitor 16.
Note: We have neglected a few amino acids of some figures for clarity images. The ligand is represented "balls and sticks" of different colors, and amino acids in the active site of the enzyme are shown in "wireframe".
The compound 16 establishes several Van Der Waals interactions with metal ions. In the Table 4, we have summarized the different interactions (Figure 2).
| N | Residues involved | Atom of amino acid | Ligand atom | Distance Å |
|---|---|---|---|---|
| 4 | CO479 | - | O4 | 2.218 |
| - | O5 | 2.352 | ||
| - | C8 | 2.613 | ||
| C12 | 2.596 | |||
| 3 | CO480 | - | O4 | 2.220 |
| - | O7 | 2.668 | ||
| - | C8 | 2.975 |
Table 4: Van der Waals interactions.
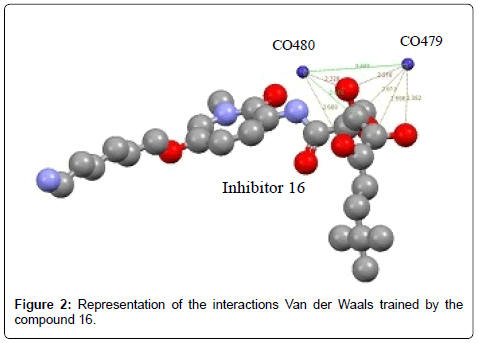
Figure 2: Representation of the interactions Van der Waals trained by the compound 16.
The results obtained in our study are consistent with those found by other authors who have shown that bengamide derivatives are powerful inhibitors of methionine aminopeptidase [18,19].
The test by the RMSD allows us to conclude that GOLD is an excellent program for molecular docking, and seems well adapted to the study of this type of theoretical calculation. The docking results show that the compound 16 represents the best inhibitor to fight the tumors. An in vitro and in vivo experimental study will allow verifying later the theoretical results obtained in silico.