Journal of Glycobiology
Open Access
ISSN: 2168-958X
ISSN: 2168-958X
Review Article - (2015) Volume 4, Issue 1
Keywords: Signaling pathways; Heparan sulfate glycosaminoglycans; ErbB3 receptor; Receptor tyrosine kinase (RTK); Cell-signaling; FGFR system
Typically, binding on monomeric ligand to monomeric receptors leads to Michaelis-Menten type activation of intracellular signaling cascades. Specifically, as the concentration of extracellular ligand increases from low to high, larger fraction of receptor gets activated until the ligand concentration becomes high enough such that receptor activation is saturated. Receptor activation initiates a cascade of enzymatic reactions that lead to phosphorylation of effector molecules like ERK and AKT amongst others. Thus, saturable activation of receptor by ligand leads to saturable activation of effector molecules. Accordingly, one would expect that cellular responses to signaling pathway activation also follow Michaelis-Menten type kinetics. This is indeed the case for signaling pathways like ErbB and IGFR at physiological ligand concentrations [1-6].
Interestingly, some cells respond to FGF signaling in an atypical fashion. Instead of following Michaelis-Menten type reaction kinetics and responding in a saturable fashion, cells respond in a biphasic manner [7-11]. Specifically, from low to intermediate concentrations of FGF-ligands, cellular response increases but then decreases from intermediate to high concentrations of FGF-ligand. For example, FGF2- induced neurite outgrowth reaches a peak at 4ng/ml and decreases for FGF2 concentrations from 10-200 ng/ml [12-14]. Similarly, FGFinduced proliferation of NIH3T3 cells also reaches a maximum at 1ng/ml of FGF2 and decreases for 10-100ng/ml concentrations [13]. Such biphasic response has been shown for fibroblasts and osteoblasts as well [14,15]. Despite multiple instances of biphasic phenotypic responses to FGFR signaling, the underlying molecular mechanism for these atypical biphasic responses remained unexplored until recently.
To address this question we have investigated the mechanistic rationale for biphasic response to FGFR signaling by specific ligands [9]. We calibrated an Ordinary-Differential-Equation (ODE)-based mathematical model to high-density signaling data (dynamic pERK response to FGF2 stimulation at 11 time points over 120 minutes) using a published particle swarm optimization algorithm combined with a
novel feature-based constraint approach [16]. The proposed model structure was based on published data including binding affinities, crystal-structures and signaling effects [17-20]. Briefly, FGF2 binds to HSGAG to form a dimer which subsequentially binds to FGFR to form a trimeric complex. The trimer dimerizes to form the signaling unit that recruits FRS2, enzymatically phosphorylates it and subsequently activates the intracellular Ras-Raf signaling cascade. The model also accounts for the negative feedback loop where downstream effector molecule pERK, binds to upstream FRS2 and pFRS2 which ultimately leads to their degradation [21]. The model was validated by testing model simulation predictions with in-vitro experiments for multiple intracellular and extracellular perturbations. We demonstrated that the reduced model accurately predicted pERK responses to multiple perturbations and thus can be considered a parsed version of the FGFRnetwork that captures the essential features of the pathway and thus can be utilized to investigate FGFR pathway behavior. It is noteworthy at this point that the quantitative relationship between biphasic signaling response and corresponding biphasic phenotypic response is currently missing. Such experimental data will be the subject of future work and can help establish a link from ligand concentration in media all the way to cellular phenotypic response.
An in-depth investigation of the model indicates that ligands like FGF2 elicit biphasic responses owing to the ternary interactions between FGF2, HSGAGs and FGFRs on the surface of the cell. Specifically, HSGAGs and FGFRs compete to bind with FGF2 at all ligand-concentrations. Furthermore, at low to intermediate FGF2- concentrations, there exist enough free FGFRs on the cell surface in addition to the FGF2-HSGAG and FGF2-FGFR complexes. Thus, FGF2-HSGAG complex can bind to free FGFRs and form the trimeric signaling unit. In contrast, at high levels of FGF2, a large fraction of FGFRs are bound to FGF2 and trimeric signaling units cannot form because binding of FGF2-FGFR complex to HSGAG is weak, thereby leading to a decrease in pERK response (Figure 1). Although the existence of biphasic response is initiated on the cell surface, the magnitude of such biphasic behavior is controlled by the intracellular signaling cascade. Thus, ternary interactions on the cell surface combine with intracellular signaling to regulate the specific cellular responses observed.
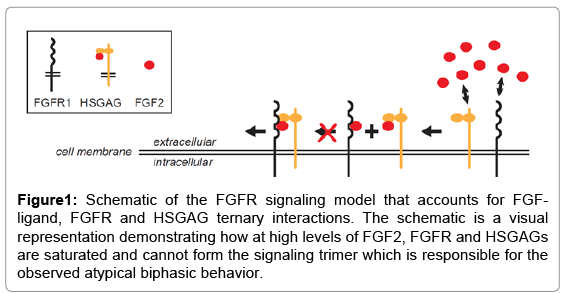
Figure 1: Schematic of the FGFR signaling model that accounts for FGFligand, FGFR and HSGAG ternary interactions. The schematic is a visual representation demonstrating how at high levels of FGF2, FGFR and HSGAGs are saturated and cannot form the signaling trimer which is responsible for the observed atypical biphasic behavior.
These results shed light on the critical role of HSGAGs in regulation of signaling pathways. They indicate that HSGAGs are not just passive scaffolds that facilitate interactions between ligands and receptors physically but actively participate in signaling response [22] HSGAGs interact with the ligands of multiple RTK families and coordinate the binding of ligands to their respective receptors. In addition, HSGAGs also stabilize the ternary complex of ligands and receptors [23]. Furthermore, targeted disruption of the HS interaction with HGF & VEGF by means of amino acid substitution on key basic residues that govern the HS-HGF & HS-VEGF surface interaction uncovered a novel mechanism of action, where the mutant proteins acted as selective competitive antagonists of their respective normal and oncogenic signaling pathways. These findings further highlight the critical role for HSPG in FGF, MET, VEGF, HB-EGF, PDGF and TGF-β RTK signaling pathways. [8,24-27].
One of the biggest hurdles in investigating the role of HSGAGs is that currently measuring HS patterns is rather difficult experimentally, requiring specialized reagents such as pattern specific HS antibodies to measure HS structures on the cell surface [28]. HSGAGs on the cell surface are not a single homogenous mixture but rather a heterogeneous mixture of molecules with different lengths and sulfation patterns. Fractions of specific species with specific lengths and patterns can bind to different ligands with different affinities and differentially modulate signaling responses [29]. Therefore, in the absence of detailed HSGAG measurements and characterization, our work provides an alternative approach for understanding the relevance of HSGAGs in the regulation of signaling responses. Of particular interest in the future, would be the integration of HS pattern analysis into the current signaling model which will help further define the exact mechanisms of receptor activation in both normal and pathologic situations.
Looking forward, this data offers a new perspective on the FGFR signaling pathway which has recently been verified as a bona-fide therapeutic target across multiple indications, including lung and breast cancer [30]. Finally, a deeper understanding of the mechanisms of signaling regulated by HSGAGs should inform better therapeutic design which might translate to improved patient outcomes. It would also be interesting to extend this model explore any differences that might exist between cellular response to endocrine vs. paracrine liganddependent activation, different FGF-ligands as well as how different cell-types with different combinations of HSGAGs would influence signaling pathways.