Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Review Article - (2013) Volume 1, Issue 1
Keywords: Chronic myeloid leukemia (CML); Philadelphia chromosome (Ph) chromosome; CML susceptibility; Genetic polymorphisms
Chronic myeloid leukemia (CML), also known as chronic myelocytic or chronic myelogenous leukemia, is a clonal myeloproliferative disorder characterized by expansion of transformed, primitive hematopoietic progenitor cells. It was first recognized as a clinical entity by John Hughes Bennett in the mid-1840s [1-4]. The myeloid progenitor cells expand in various stages of maturation, being released into the peripheral blood and subsequently following to extramedullary sites. The disorderly sprawl of these cells reflects the occurrence of alterations in their proliferative capacity, as well as changes in the balance between self-renewal and differentiation, increasing the number of transformed cells, and reducing the number of pluripotent stem cells [1,4].
CML can be classified into three disease phases: chronic phase (CP), accelerated phase (AP), and terminal blast phase (BP). Approximately 90% of patients are diagnosed during the CP, although in this phase 20% to 40% of patients may remain asymptomatic [3,5-7]. Common symptoms of CML in CP, when present, are generally related to the expansion of CML cells, and mainly include fatigue, weakness, headaches, weight loss, malaise, easy satiety and discomfort or left upper quadrant pain, caused primarily by anemia and splenomegaly [3,6], although most patients do not present anemia in the diagnosis and, in general, their quality of life is not altered in this phase, especially if the leukocyte count is controlled [8]. Most patients have basophilia and eosinophilia [7]. Neutrophilic leukocytosis is also a common feature of CP [9], and the white blood cell (WBC) count can be as high as 100,000 cells/mL, leading in rare instances to signs and symptoms of hyperviscosity, such as retinal hemorrhage, priapism (usually with marked leukocytosis or thrombocytosis), cerebrovascular accidents, upper gastrointestinal ulceration and bleeding (from elevated histamine levels due to basophilia). Also common is the occurrence of platelet dysfunction [3,6].
Before introduction of tyrosine kinase inhibitors (TKI) (1973- 2000) [10], the median survival of patients with CML after diagnosis was 4-6 years, and the stages following the CP had a short duration, corresponding to the terminal period of the disease [8], with AP lasting 4 to 6 months and BP, a few months [11]. Nowadays (post TKI era), the median survival of patients has increased to 13-15 years for CP, and 6-12 months for both AP and BC [12,13]. However, although progression through all stages is most common, the time course for progression can be extremely varied, and 20% to 25% of patients progress directly from CP to BP [5].
CML constitutes about 15-20% of all newly diagnosed cases of leukemia in adults and occurs with an incidence of approximately 1-2 cases in 100,000/year, with an estimated survival rate of 90% at 5 years and an annual mortality rate of 2% [3,6,7,9,14]. The median age of onset of CML is about 40-60 years, with less than 10% of the cases in patients aged less than 20 years [7,9,15]. It is more common among men than women, with a male-to-female ratio of 1.4 to 2.2:1 [16]. Although the clinical course has been described as being similar in both sexes [16], a recent analysis performed by Mandal et al. [10] from the Surveillance Epidemiology and End Results of the US National Cancer Institute (SEER) to see survival differences from the pre-TKI (1973-2000) to post-TKI eras (2002-2008) showed that, although the survival rates of the older population were lower than the younger population for each group (for both pre and post-TKI era), and there was a relative survival increase from the pre- to post-TKI era for all ethnic groups, the survival rate of young African-American men (<50 years) was significantly lower compared to young Caucasian men in the pre-TKI era, while young African-American women (<50 years) with CML had lower relative survival rates compared to young Caucasian women in the postimatinib era. According to the authors, while socioeconomic status and access to healthcare have been known to impact survival in the past, which could explain the lower survival in African-American men that we observed in the pre-TKI era, in the post-TKI era higher rates of imatinib resistance in the African-American group could explain the lower relative survival rates observed for African-American women [10]. This means that at least among young women with CML using TKI, the ethnic differences should be taken into account in association studies involving CML. Also for men <50 years, such ethnic differences take into account underdeveloped and developing countries, where socioeconomic status and access to healthcare still can cause an impact on survival.
The etiology of CML is related to a chromosomal translocation, discovered by Nowell and Hungerford, on observing an anomalous chromosome of group G [17], the Philadelphia chromosome, often abbreviated as Ph, Ph(1), or Ph1, and present in 95% of individuals affected [4,18]. With the improvement of chromosome banding techniques, Rowley [19] described the chromosomal abnormality that exists in these patients with CML as a reciprocal translocation between two chromosomes, where chromosome 22 shows loss of the terminal portion of its long arm and chromosome 9 presents a gain of this genetic material in the terminal portion of its long arm.
The classic definition of Ph chromosome is t(9;22)(q34;q11), which indicates that this chromosome results from a reciprocal translocation of cytogenetic material following a break on chromosome 9 at band q34, and a break on chromosome 22 at band q11 [1,4,5,9,20,21]. As a result of these breaks, the 3’ part of the ABL1 (Abelson murine leukemia viral oncogene homolog 1) gene is moved from chromosome 9 (its normal locus) to chromosome 22, and is juxtaposed to a segment of the BCR (Breakpoint Cluster Region) gene on chromosome 22. The BCR gene is also disrupted, and its 3’ end is moved from chromosome 22 to chromosome 9, while its 5’ domain remains on chromosome 22 [6,9]. Thus, the Ph chromosome has a chimeric gene resulting from the fusion of the 5’ end closer to the centromere of the BCR gene on chromosome 22q11.23, with the 3’ end of the exon 2 (known as a2) of the ABL1 protooncogene, located at chromosome 9q34.1 (Figure 1). This configuration places the fusion gene under the control of the BCR promoter [1,9,20]. The hybrid BCR/ABL oncogene encodes a constitutively active BCR-ABL fusion tyrosine-kinase protein, which has a direct and crucial participation in the development of CML, by stimulating uncontrolled proliferation of transformed cells, alteration in the balance of self-renewal/differentiation (discordant maturation) and in cell adhesion properties, and escape from apoptosis, generating genomic instability and leading to mutations and chromosomal abnormalities [4,6,20–24].
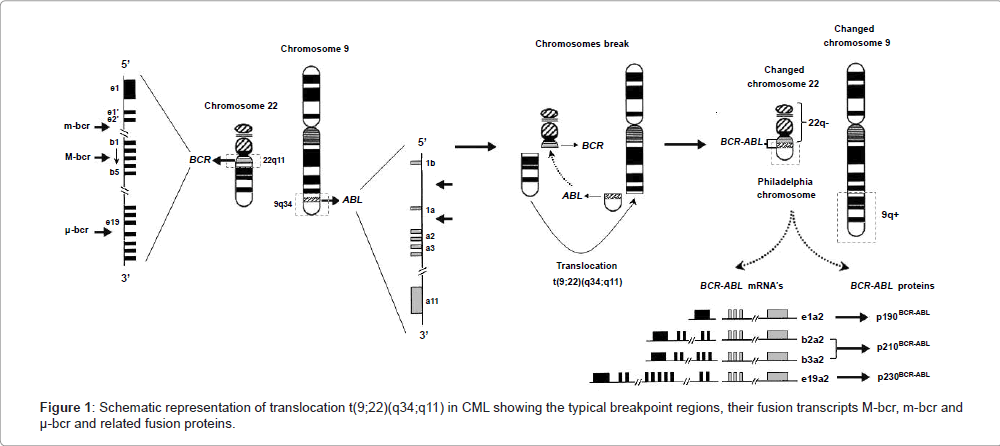
Figure 1: Schematic representation of translocation t(9;22)(q34;q11) in CML showing the typical breakpoint regions, their fusion transcripts M-bcr, m-bcr and μ-bcr and related fusion proteins.
It is known that the variation of breakpoints of the BCR gene involved in translocation between chromosomes 9 and 22 can promote the formation of different transcripts of BCR/ABL genes and their products, but all BCR/ABL1 fusion genes contain a variable 5’ portion derived from BCR sequences and a 3’ portion almost invariably of the ABL1 gene sequence [25,26]. Heisterkamp et al. [27] determined the structural organization of the BCR gene, which contains 23 exons and occupies a region of about 130 kb on chromosome 22. In the vast majority of CML patients, the break on chromosome 22 involves an area of 5.8 kilobases (kb) termed the major breakpoint cluster region (M-bcr), which contains five exons corresponding to exons 12 to 16, originally numbered from b1 to b5 [26]. The breaks occur within introns located downstream of either exon 13 (known as e13 or b2) or 14 (known as e14 or b3) with the introns upstream of ABL1 exon 2 (a2) forming the fusion gene b2a2 or b3a2, respectively. This rearrangement generates a BCR/ABL1 hybrid gene that is expressed as an 8.5 kb chimeric mRNA with a b2a2 or b3a2 junction, and translated into a 210 kilodaltons (kDa) fusion protein (p210BCR-ABL), which is associated with the underlying mechanism in the chronic phase of CML [4,20,26,28-30]. The breakpoint in the BCR gene is also found within the M-bcr region (p210) in 30% of adults and 20% of children with Philadelphia chromosome-positive (Ph+) Acute Lymphoblastic Leukemia (Ph+ ALL), as well as in patients with Acute Myeloid Leukemia (AML) [31-35]. There are also two less common breakpoints in the intronic region between the alternative BCR exon 2 known as minor breakpoint cluster region (m-bcr), and between BCR exons 19 and 20, known as microbreakpoint cluster region (μ-bcr), which encode, respectively, a 190-kDa (p190BCR-ABL, e1a2 transcripts) and a 230-kDa (p230BCR-ABL, e19a2 transcripts) fusion protein (Figure 1) [28,29]. Transcript e1a2 (p190) rarely occurs in CML patients and it has been associated with an inferior outcome to therapy with tyrosine kinase inhibitors, but it is commonly found in Ph+ ALL patients and occasionally in AML. Junctions e19 and e20 (p230) also rarely occur in CML or in a clinical entity called neutrophilic CML; the latter has been reported to have a more benign clinical course than that associated with the traditional b2/a2 or b3/a2 fusion (p210) [31,32,36].
Although 5% of cases of CML do not exhibit the Ph chromosome in classical cytogenetic analysis, molecular analysis detects BCR/ABL rearrangements in most cases [18]. As patients progress through the different phases, a distinct feature of disease progression is the appearance of additional cytogenetic abnormalities in the Ph+ cells [5]. This phenomenon, known as clonal evolution, frequently involves a second Ph (30%), trisomy of chromosome 8 (33%), isochromosome 17 (20%), trisomy of chromosome 21 (7%) and monosomy of chromosome 7 (5%), besides other chromosome abnormalities [37–39]. Mutations and deletions may also occur in specific genes that regulate the cell cycle (eg. INK4A, ARF, p16/INK4a, CDKN2A-CDKN2B, p53 and RB) [1,5,18,40,41], as well as in IKAROS and PAX5 genes [37], required for normal lymphoid development [41,42], thereby facilitating leukaemiainitiating cell self-renewal and enhancing targeted therapeutic resistance in vivo [40]. Thus, as previously mentioned, the BCR/ABLtransformed cell seems endowed with an intrinsic genetic instability, which leads to a progressive accumulation of genetic lesions. This progression represents the natural end of all CML cases and the main cause of death in CML patients [18].
The proteins generated from different breakpoints have been the subject of a number of studies, many of which, seeking a better understanding of CML, focused on investigating the physiological function of the proteins encoded by the ABL1 and BCR genes.
Proteins encoded by the normal ABL1 and BCR genes
The normal ABL1 gene encodes the c-ABL1 protein (145 kDa), which belongs to the Scr family of non-receptor tyrosine kinases. This protein is ubiquitously expressed in the cell membrane, actin cytoskeleton, cytosol and nucleus, regulating various cellular processes, such as mitogenesis, migration, adhesion, response to DNA damage, survival and response to oxidative stress [43]. The structural organization of c-ABL1 domains is similar to the Src protein kinase (Avian Sarcoma), having SH1 tyrosine kinase (Src Homology 1), SH2 and SH3 domains toward the N-terminus. At the C-terminal end, the c-ABL molecule has a sequence rich in proline (PXXP), which interacts with SH3 domains of adapter proteins; a DNA binding site; a binding site for both F-actin and G-actin; a nuclear localization signal (NLS); and a nuclear export signal (NES) (Figure 2) [44]. Because of its potentially deleterious effects, the tyrosine kinase activity of c-ABL1 must be very tightly controlled in the cell. This is achieved by autoinhibition through a complex set of intramolecular interactions that involve the SH3 and SH2 domains, the catalytic tyrosine kinase domain, and all other segments in the aminoterminal half of the protein. The autoinhibited form of c-ABL1 is not phosphorylated on tyrosine residues [45].
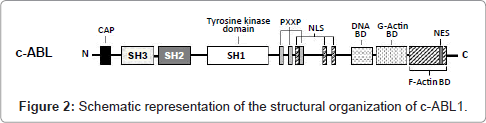
Figure 2: Schematic representation of the structural organization of c-ABL1.
The BCR gene is also expressed ubiquitously, and encodes a 1271 amino acid cytoplasmic phosphoprotein of 160 kDa molecular mass, found primarily in the brain and hematopoietic cells at early stages of myeloid differentiation, and whose levels are significantly reduced in mature polymorphonuclear leukocytes [4,20]. In the first exon-encoded sequences (amino acids 1-427), BCR contains a coiled-coil domain that mediates homo-oligomerization, and a domain of serine/threonine kinase activity containing SH2-binding domains (noncatalytic regions of ~100 amino acids that bind SH2-binding sites), where other proteins can bind to the phosphoamino acid serine, threonine, or tyrosine (rather than only tyrosine). This interaction is important in the assembly of signal transduction [4,20,46]. A consensus sequence homologous to the ATP-binding site of other kinases as well as a likely phosphotransferase domain has also been identified, allowing BCR to autophosphorylate on serine and threonine residues, as well as to transphosphorylate casein and histones (known substrates for serine/threonine kinases) in vitro [20]. The central region of the BCR protein (amino acids 490 to 690; encoded by exons 3 to 10) contains a region of homology to the DBL oncoprotein (with an associated pleckstrin homology domain) [4,46] that functions as a guanine nucleotide-exchange factor (GEF, that activates G proteins) for RHO proteins [4], a family of GTPases that has been intensively studied for their roles in signal transduction processes which lead to cytoskeletal-dependent responses, including cell migration and phagocytosis, besides being important regulators of cell cycle progression and affecting the expression of a number of genes, including those for matrix-degrading proteases implicated in cancer invasion [47]. This region can interact with the protein Xeroderma Pigmentosum group B protein (XPB), which plays an important role in DNA repair and cell cycle regulation [20,48]. The C-terminus of BCR, in turn, contains a domain with GTPase activating protein (GAP, which inactivates G proteins) homologous to the GAP protein for p21RHO (RhoGAP), which is involved in the regulation of the actin cytoskeleton, having GAP activity toward the Ras-related GTP-binding protein (p21RAC) and Cdc42 proteins (Figure 3) [4,20,49]. Although the normal function of the BCR gene product is not known and it has no intrinsic oncogenic properties [4], these data indicate that BCR has both GEF and GAP functions, suggesting a dual role for this molecule in G protein-associated signaling pathways, besides implicating the participation of BCR in two major intracellular signaling mechanisms in eukaryotic cells: phosphorylation and GTP-binding [20].
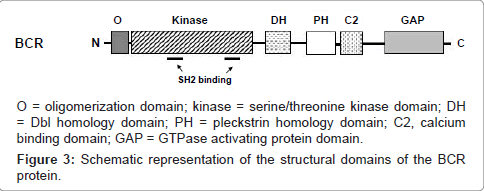
Figure 3: Schematic representation of the structural domains of the BCR protein.
BCR-ABL1 fusion proteins
The three principal forms of BCR-ABL1 fusion proteins (p190, p210, and p230) contain almost all c-ABL1 (except the first exonencoded sequence), including the entire ABL tyrosine kinase catalytic domain, but they have different amounts of BCR sequences: only p210 and p230 include the DBL/plekstrin homology domains from the central portion of BCR, and a portion of C-terminal GAP domain of BCR is retained only in the longer p230 fusion protein (it is not included in p190 and p210). However, the first exon-encoded sequence of the BCR gene is the one included in all known BCR-ABL fusion proteins (Figure 1) [4,20,31]. As mentioned above, within this region of BCR resides an oligomerization domain and the kinase domains containing SH2-binding domains [4,20,46]. The last allows the binding of BCR to the c-ABL1 SH2 sequences in the phosphoamino acids serine and threonine (but not phosphotyrosine residues on BCR) [20,50]. The BCR sequences involved in this interaction, which lie between amino acids 192-242 and 298-413, are essential for the activation of the c-ABL1 tyrosine kinase, and thus, for the oncogenic activation of BCR-ABL [20,46,50]. This occurs because the oligomerization domain of BCR mediates dimerization and/or tetramerization of BCR-ABL, allowing a phosphorylation activated by loop that stabilizes a conformation compatible with substrate binding and catalysis, stimulating self-phosphorylations [45]. Thus, all the three BCR-ABL isoforms have increased tyrosine kinase relative to c-ABL1, due to the addition of BCR exon 1 sequences to c-ABL1 [4]. BCR-ABL creates a constitutively active tyrosine kinase which activates several signal transduction pathways from the cytoplasm to the nucleus [51,52], which are also utilized by hematopoietic growth factors, including steel factor, thrombopoietin, interleukin-3, and granulocyte/macrophagecolony stimulating factor [52].
BCR-ABL signaling prevents down-regulation of cyclin-dependent kinase activity and cell cycle arrest after growth factor deprivation of hematopoietic progenitor cells, its expression being sufficient to induce G1-to-S phase transition, DNA synthesis, and activation of cyclindependent kinases in cells that were arrested in G0 by growth factor deprivation [53], besides inhibiting apoptosis through activation of a Ras-dependent signaling pathway [54]. Thus, the primary mitogenic activity of BCR-ABL, on stimulating the transformed hematopoietic growth factor-dependent cell lines to enter in the cell cycle, leads to growth factor independence [52,53].
Considering the primary structures of p190, p210, and p230, it would be plausible to suppose that P210 would have a higher tyrosine kinase activity due to the DBL/plekstrin homology domains which function as a GEF (that activates G proteins), and, thus, a higher oncogenic activity than p190, which in turn would have larger oncogenic activity than P230 (due to a portion of a GAP catalytic domain, which inactivates G proteins). However, as revised by Van Etten [4], p190 has been reported as having the highest intrinsic kinase activity, followed by p210 and p230, as well as being a parameter of CML relapse [55]; it is also associated with an inferior outcome to therapy with TKI and with high-risk patients [36]. A possible explanation would be that the signal transducer and activator of transcription STAT6, a transcription factor normally activated by IL-4 and implicated in lymphoid proliferative responses, is preferentially tyrosine phosphorylated and activated by p190 (but not by p210) [31].
Although the clinical and biological aspects of CML are well documented, little is known about individual susceptibility to this disease [24]. There are no known hereditary, familial, geographic, ethnic, or economic associations with CML [3], and the mechanisms behind disease progression are not fully understood [5]. However, while a specific chromosomal translocation is the common oncogenetic mechanism of CML, it may have environmental causes such as irradiation [3,56] and chemical exposure to benzene [57-59] and other xenobiotics [58,60-62]. In most patients, the factor responsible for the induction of the Ph chromosome is unknown, although CML has been observed with increased frequency in individuals exposed to the atom bomb explosions of 1945 in Japan, in radiologists, and in patients with ankylosing spondylitis treated with radiation therapy [3], as well as still being diagnosed together with other oncohematological disorders in Ukrainian clean-up workers 10-18 years after the Chernobyl accident [56]. Moreover, benzene is a known human carcinogen, with a substantial number of case reports and epidemiological studies providing evidence of a causal relationship between occupational (chronic) exposure and various types of leukaemia [57,59]. An increased risk of CML has been reported after exposure to benzene, petrol refining products, polycyclic aromatic hydrocarbons, and electromagnetic fields among men [58], while a meta-analysis has confirmed an increased risk of CML related to pesticide applicators among men and farmers or agricultural workers [60], and associations between childhood leukemia with both parental occupational and residential pesticide exposure have been reported in review articles [61,62].
Oxidative stress, a state generated by an excessive production of reactive oxygen species (ROS) and/or by a deficiency in antioxidant pathways, has also been observed in several hematopoietic malignancies, including acute and chronic myeloid leukemias [63]. It is known that BCR/ABL expression stimulates ROS production in hematopoietic progenitor cells [64], and studies have suggested that ROS may contribute to increased DNA damage in CML cells [22]. Leukemia cell lines expressing BCR-ABL1 kinase accumulate ROS and oxidative DNA damage, resulting in genomic instability, which leads to disease relapse and/or malignant progression to a fatal blast phase [65]. These data suggest a probable genetic susceptibility to this disease, related to the body’s ability to defend itself from oxidative stress and/or external aggression such as chemicals, ionizing radiation and others, through functional polymorphic variant enzymes that metabolize toxicants and/or protect against oxidative stress. Thus, association studies have been performed to identify genetic variants associated with CML susceptibility, and among them are the polymorphisms in the genes of methylenetetrahydrofolate reductase (MTHFR) [24,66-74], glutathione S-transferases (GSTs M1 and T1) [24,74-87], and haptoglobin (Hp) [24,74,88-96], among others that will not be covered in this review.
Considering that: (1) as previously presented, it has been showed that there are differences among CML survival rates pre- and post- TKI by gender and race/ethnicity [10]; (2) the age-adjusted incidence and death rates of CML based on cases diagnosed in 2005-2009 in the U.S. population geographic areas also showed differences by gender and race/ethnicity as showed in table 1 [13], and this program is a population-based cancer register covering more than 25% of the U.S. population across several disparate geographical regions [10]; and (3) MTHFR, GSTs M1 and T1 and Hp polymorphisms vary among different ethnic groups as can be seen in tables 2-4; although we intend to outline the reasons for the possible influence of these polymorphisms on the susceptibility to CML and present the association studies related to them, studies in larger populations of patients and controls of both genders should be conducted in populations worldwide to better understand the biological significance of these polymorphisms in CML susceptibility in each ethnic group and by gender, as well as in understanding their role in the variability of the disease course and in responsiveness to therapeutic agents. This is because the conflicting results among the various association studies carried out in different ethnic groups focusing on the issue could be not due to ethnic differences but instead the result of methodological problems: several studies have analyzed relatively small groups of patients and controls, resulting in very broad intervals of statistical confidence of relative risks. Thus, although the influence of these polymorphisms in the susceptibility of CML is very plausible, worldwide studies involving larger populations of patients and controls by gender and race/ethnicity are necessary to avoid any doubt about the role of these polymorphisms in CML.
| Race/Ethnicity | Male | Female | ||
| Incidence Rates (per 100,000 men) | Death Rates (per 100,000 men) | Incidence Rates (per 100,000 women) | Death Rates (per 100,000 women) | |
| White | 2.1 | 0.4 | 1.2 | 0.2 |
| Black | 1.9 | 0.5 | 1.2 | 0.3 |
| Asian/Pacific Islander | 1.5 | 0.2 | 0.7 | 0.1 |
| American Indian/Alaska Native | 1.7 | 0.6 | * | * |
| Hispanic | 1.7 | 0.3 | 1.1 | 0.2 |
Table 1: The age-adjusted incidence and death rates of CML based on cases diagnosed in 2005-2009 in the U.S. population, by gender and race/ethnicity according to the Surveillance Epidemiology and End Results (SEER) of the National Cancer Institute [13].
DNA methylation, an essential epigenetic mechanism that plays critical roles in gene expression regulation, cell differentiation and genomic integrity, is catalyzed by methyltransferases, which use the universal methyl donor S-adenosyl-L-methionine (SAM) [97]. Methylenetetrahydrofolate reductase (MTHFR, EC 1.5.1.20), a key enzyme in homocysteine (Hcy) and folate metabolism, plays an important role in DNA methylation and provision of nucleotides for DNA synthesis [24]. MTHFR catalyzes the irreversible conversion of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate, the methyl donor for synthesis of methionine from homocysteine. Methionine in turn, is converted to SAM (Figure 4), which methylates specific cytosines in DNA, regulating gene transcription through methylation [24,97,98]. Because DNA hypomethylation has been linked to abnormal chromosome rearrangements [99], and methylation abnormalities also play a role in chromosome segregation processes in cancer cells [100], a decrease in MTHFR activity which results in hypomethylation may leads to alteration in chromosome recombination and abnormal chromosome segregation [101].
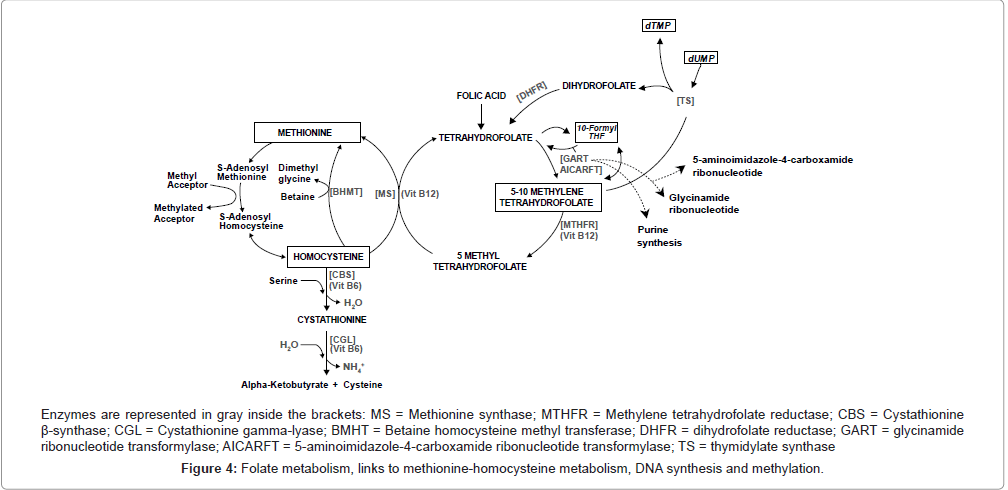
Figure 4: Folate metabolism, links to methionine-homocysteine metabolism, DNA synthesis and methylation.
On the other hand, the substrate of MTHFR, 5,10-methylenetetrahydrofolate, is also the cofactor for thymidylate synthase and is required for the production of deoxythymidine monophosphate (dTMP) via this enzyme [102]. The 5,10-methylenetetrahydrofolate donates a methyl group to deoxyuracil monophosphate (dUMP) converting it to dTMP (Figure 4), which is used for DNA synthesis and repair [103]. So, if folate is continually limited, low cytosolic levels of 5,10-methylenetetrahydrofolate decrease synthesis of dTMP, increasing the cellular ratio of dUMP/dTMP and thus the misincorporation of dUMP instead of dTMP during DNA replication [102,103]. As a result, the excision of uracil from DNA by uracil-DNA glycosylase and apyrimidinic endonuclease generates transient single-strand breaks (nicks) that could result in a less reparable and more hazardous double-strand break if two opposing nicks are formed [102]. This catastrophic repair cycle could lead to double-strand breaks and chromosomal damage [103], contributing to the increased risk of chromosomal aberrations and presumably the onset of the leukemogenic process associated with folate deficiency in humans [69,102,104]. Therefore, MTHFR also plays a role in providing nucleotides essential for DNA synthesis [105], because under situations of lower MTHFR activity, more 5,10-methylenetetrahydrofolate will be available for dTMP synthesis when in situations of folate deficiency status, preventing imbalances of nucleotide pools during DNA synthesis [24,106]. Thus, polymorphic variants of MTHFR that lead to enhanced dTMP pools and better quality DNA synthesis could promote some protection from the development of leukemia. This is particularly true of variants with translocations, but only if the activity of MTHFR is not reduced enough to cause DNA hypomethylation, and if the MTHFR variant does not affect plasma folate levels, which could lead to uracil misincorporation into DNA, especially in individuals with low folate intake.
In view of all the explanations, mutations in the MTHFR gene related to the decrease in MTHFR activity might explain the interindividual differences in the increase of chromosome breakage and damage. Therefore, the MTHFR gene is a good candidate for CML etiology [101].
The MTHFR gene is located on the short arm of chromosome 1 (1p36.3), and its complementary DNA (cDNA) sequence, which includes 11 exons ranging in size from 103 base pairs (bp) to 432 bp, is approximately 2.2 kb [107,108]. Several mutations in the MTHFR gene have been identified, but most are rare and associated with severe enzymatic deficiency [107]. However, in contrast with these rarities, two common thermolabile single-nucleotide polymorphisms (SNPs) in the MTHFR gene (C677T and A1298C) deserve special mention in CML susceptibility because they result in a decrease in MTHFR activity [109,110], and its possible consequent effects mentioned above.
It has been shown that the SNPs C677T (dbSNP rs1801133) and A1298C (dbSNP rs1801131) in the MTHFR gene are involved in increased meiotic chromosomal non-disjunction and risk of some aneuploidies, mainly Down’s syndrome [111-118]. Given that recurrent chromosomal abnormalities (such as a second Ph chromosome) added to new structural chromosome abnormalities and aneuploidies are important factors in the clonal evolution and pathogenesis of CML, where a large number of chromosome abnormalities are associated with the morphology and clinical subsets of CML, as well as its prognosis [37-39,101], the MTHFR C677T and A1298C polymorphisms are good candidates not only for CML etiology, but also for the chromosome abnormalities associated with the clonal evolution of CML, and thus for the disease progression and its consequent effects.
The transition C→T at nucleotide 677 (C677T; dbSNP rs1801133) in exon 4 of the MTHFR gene, resulting in the substitution of an alanine (GCC) for a valine (GTC) at position 222 (Ala222Val) in the N-terminal catalytic domain of the protein, which generates a thermolabile variant of the enzyme was first identified by Frosst et al. [98,108-110,119]. The 677TT homozygosis has been associated with reduced MTHFR activity and plasma folate levels, as well as elevated plasma Hcy concentrations (mild to moderate hyperhomocysteinemia), especially in patients with low folate levels [98,101,120]. It has been reported that MTHFR activity is reduced to 70% in homozygous TT and 30-40% in heterozygous CT with regard to the normal mean value [101,109,121]. Also, serum folate concentration is lower in individuals with the MTHFR 677TT genotype than in those with the MTHFR 677CC or 677CT genotypes [122]. Therefore, decrease in the MTHFR activity and serum folate levels in individuals carrying the MTHFR 677TT genotype could affect genomic stability by diminishing DNA methylation and favoring uracil misincorporation into DNA with its consequent double-strand breaks induced during DNA repair, mainly in condition of low folate intake.
The frequency of the MTHFR C677T polymorphism varies among ethnic groups. The C677T polymorphism is found in approximately 12% of the general population, with variations from 5% to 18% [98]. Analysis of Caucasian and Asian populations typically shows rates of ~12% for homozygous T/T and up to 50% for heterozygous C/T. African-Americans exhibit very low incidence of T/T genotype, whereas European Caucasians exhibit substantial variation [123].
The second polymorphism of the MTHFR gene, A1298C (dbSNP rs1801131), which occurs in exon 7, resulting in the replacement of glutamate (Glu or E) with alanine (Ala or A) at position 429 of the enzyme (Glu429Ala or E429A), was first identified by Van der Put et al. (1998) [108,110,124]. Since it lies in the SAM-regulatory domain of the enzyme (within the C-terminal regulatory domain) [24,98,108,124], the binding of SAM results in conformational changes within the MTHFR enzyme that inhibit the enzyme’s activity [108]. Thus, folate intake may affect MTHFR gene expression, regulating cellular SAM levels [98].
Although the MTHFR A1298C polymorphism has been observed in approximately 10% of individuals and is associated with reduced enzyme activity, it alone does not lead to a severe enzyme deficiency and does not seem to be associated with reduced plasma folate levels or elevated Hcy [98]. On the other hand, the combination of heterozygosis of both polymorphisms of MTHFR (C677T and A1298C) leads to features similar to those observed in homozygotes 677TT, and has been associated with reduced enzyme activity, decreased plasma folate levels and hyperhomocysteinemia [98,110].
Association studies between the MTHFR polymorphisms C677T and A1298C and risk of CML have been carried out in different populations, but several of them with conflicting results, as shown in table 2.
Although some of these studies were carried out with small samples mainly consisting of patients, where the previously mentioned methodological problems cannot be discarded, these conflicting results may also be due to: (1) studies considering folate intake are limited [122]; (2) individuals with the MTHFR 677TT genotype may need to consume more folate to maintain serum folate levels similar to those found in individuals with the 677CC/677CT genotypes [122]; and (3) the MTHFR A1298C polymorphism alone does not lead to a severe enzyme deficiency or reduced plasma folate levels [98,110]. Thus, studies involving serum folate and MTHFR C677T and A1298C polymorphisms adjusted for folate intake are needed to better understand the biological significance of these polymorphisms in CML susceptibility, as well as in understanding the variability of the disease course and the patients’ response to medication. Moreover, since these polymorphisms vary among ethnic groups (Table 2) and populations worldwide vary considerably in their predisposition to diseases and in the allele frequencies of pharmacogenetically important loci, probably due to genetic drift or adaptation to local selective factors such as climate and available nutrients [125], studies dealing with a large population in several ethnicities are also required to reach this understanding.
| Authors, year | Ethnicity | Polymorphisms | Characteristics of cases | Characteristics of controls | Polymorphisms | Case | Control | OR (95% CI) | P value |
| Robien et al., 2004 [66] | Caucasians | MTHFR C677T A1298C | 336 CML patients The majority (86%) of the study population had a diagnosis of CML in chronic phase at the time of HCT. | – | MTHFR C677T | ||||
| 677CC | 149(44) | ||||||||
| 677CT | 137(41) | ||||||||
| 677TT | 50(15) | – | – | – | |||||
| MTHFR A1298C | |||||||||
| 1298AA | 155(46) | ||||||||
| 1298AC | 137(41) | ||||||||
| 1298CC | 44(13) | ||||||||
| Hur et al., 2006 [67] | East Asian | MTHFR C677T A1298C | 40 CML patients | 200 healthy individuals without any clinical disorder as normal controls | MTHFR C677T | ||||
| 677CC | 13(32.5) | 80(40) | 1.0 | – | |||||
| 677CT | 17 (42.5) | 80(40) | 1.31 (0.60-2.87) | – | |||||
| 677TT | 10(25) | 40(20) | 1.54 (0.62-3.81) | – | |||||
| MTHFR A1298C | |||||||||
| 1298AA | 31(77.5) | 116(58) | 1.0 | – | |||||
| 1298AC | 7(17.5) | 78(39.0) | 0.34 (0.14-0.80) | – | |||||
| 1298CC | 2(5.0) | 6(3.0) | 1.25 (0.24-6.49) | – | |||||
| Moon et al., 2007 [68] | East Asian | MTHFR C677T A1298C | 115 patients with CML (75 males, 40 females; mean age: 43.8 years | The control subjects included 434 healthy individuals (195 males, 239 females; mean age: 47.3 years; | MTHFR C677T | ||||
| 677CC | 43(37.4) | 144(33.2) | 1.00 | – | |||||
| 677CT | 45(39.1) | 196(45.2) | 0.77 (0.48-1.23) | 0.27 | |||||
| 677TT | 27(23.5) | 94(21.7) | 0.96 (0.56-1.66) | 0.89 | |||||
| MTHFR A1298C | |||||||||
| 1298AA | 74(64.3) | 307 (70.7) | 1.00 | – | |||||
| 1298AC | 33(28.7) | 120(27.6) | 1.11 (0.70-1.78) | 0.65 | |||||
| 1298CC | 8(23.5) | 7 (1.6) | 5.12 (1.75-14.9) | 0.003 | |||||
| Barbosa et al., 2008 [69] | Mixed | MTHFR C677T A1298C | 27 CML patients (15 female (53.6%) and 13 male (46.4%); median age 27 yrs | The control group consisted of 100 (47% female and 53% male; median age 29 yrs. | MTHFR C677T | ||||
| 677CC | 46(68.7) | 65(65) | 1 | – | |||||
| 677CT | 19(28.3) | 29(29) | 0.97 (0.46- 2.03) | – | |||||
| 677TT | 2(3) | 6(6) | 0.48 (0.07- 2.76) | – | |||||
| MTHFR A1298C | |||||||||
| 1298AA | 41(61.1) | 63(63) | 1.0 | – | |||||
| 1298AC | 23(34.3) | 32(32) | 1.11 (0.55- 2.25) | – | |||||
| 1298CC | 3(4.6) | 5(5) | 0.89 (0.16- 4.48) | – | |||||
| Kim et al., 2009 [70] | Korea | MTHFR C677T A1298C | 149 CML patients (94 men and 55 women; mean age 50.4±17.1 years | The control group (n = 1700 48.8% of the eligible subjects; 821 men and 879 women), mean age 52.2±14.3 years, underwent clinical examinations. | MTHFR C677T | ||||
| 677CC | 54(36) | 540(32) | 1 | – | |||||
| 677CT | 72(47) | 863(51) | 0.81 (0.55-1.17) | 0.26 | |||||
| 677TT | 26(17) | 297(17) | 0.90 (0.55-1.47) | 0.67 | |||||
| MTHFR A1298C | |||||||||
| 1298AA | 97(64) | 1147(68) | 1 | – | |||||
| 1298AC | 49(33) | 500(29) | 1.15 (0.80-1.66) | 0.44 | |||||
| 1298CC | 5(3) | 53(3) | 1.11 (0.43-2.85) | 0.83 | |||||
| Ismail et al., 2009 [71] | Jordan | MTHFR C677T A1298C | 149 patients diagnosed with CML were collected | The control group consisted of 170 healthy individuals. | MTHFR C677T | ||||
| 677CC | 63 (42.2) | 94 (55.3) | 1 | – | |||||
| 677CT | 67 (45.0) | 66 (38.8) | 1.52 (0.95-2.41) | 0.081 | |||||
| 677TT | 19 (12.8) | 10 (5.90) | 2.84 (1.24-6.50) | 0.014 | |||||
| MTHFR A1298C | |||||||||
| 1298AA | 59 (39.6) | 76 (44.7) | 1 | – | |||||
| 1298AC | 68 (45.6) | 81 (47.65) | 1.08 (0.68-1.73) | 0.743 | |||||
| 1298CC | 22 (14.8) | 13 (7.65) | 2.18 (1.01-4.69) | 0.046 | |||||
| Vahid et al., 2010 [72] | Iran | MTHFR C677T A1298C | MTHFR C677T | ||||||
| 677CC | 24 (63.15) | 56 (57.73) | 1 | – | |||||
| 677CT | 11 (28.94) | 37 (38.14) | 0.7 (0.3-1.6) | NS | |||||
| 677TT | 3 (7.89) | 4 (4.12) | 1.75 (0.36-8.4) | NS | |||||
| MTHFR A1298C | |||||||||
| 1298AA | 12 (31.57) | 39 (40.2) | 1 | – | |||||
| 1298AC | 19 (50) | 36 (37.11) | 1.7 (0.7-4.0) | NS | |||||
| 1298CC | 38 CML patients (male/female: 1.05, mean age 45.0 years, SD ± 16.7) | 97 healthy age- and sex-matched individuals (male/female: 0.94, mean age 44.8 years) participated in this experiment as the control group. | 1.03 (0.4-3.0) | NS | |||||
| Hussain et al., 2012 [73] | Indian | MTHFR C677T | MTHFR C677T | ||||||
| 677CC | 1 | – | |||||||
| 677CT | 0.84 (0.36-0.94) | 0.689 | |||||||
| 677TT | 4.5 (1.58-12.7) | 0.004 | |||||||
| Lordelo 2011 [74]; Lordelo et al., 2012 [24] |
Mixed | MTHFR C677T A1298C | 105 CML patients | The control group consisted of 273 non-related healthy volunteers of both genders. | MTHFR C677T | ||||
| 677CC | 0.741 (0.47-1.17) | 0.193 | |||||||
| 677CT | 1.130 (0.72-1.78) | 0.597 | |||||||
| 677TT | 43 CML patients | 251 healthy controls | 1.725 (0.81-3.69) | 0.156 | |||||
| MTHFR A1298C | |||||||||
| 1298AA | 1.794 (1.14-2.83) | 0.011 | |||||||
| 1298AC | 0.630 (0.40-0.99) | 0.047 | |||||||
| 1298CC | 1 (1) | 11 (4) | 0.229 (0.29-1.80) |
Table 2: Association studies between methylenetetrahydrofolate reductase (MTHFR) C677T and A1298C polymorphisms in CML patients and controls.
The glutathione S-transferases (GST; EC 2.5.1.18) are a family of enzymes ubiquitously distributed in nature that play a vital role in phase 2 of biotransformation, being responsible for the metabolism of a broad range of xenobiotics and carcinogens. These enzymes catalyze reactions involving the conjugation between reduced glutathione (GSH) and a variety of compounds containing an electrophilic center; most of them being xenobiotics or compounds endogenous to the organism (reactive oxygen metabolites) [126-131]. In addition to the conjugation reactions, various GST isoenzymes exhibit other GSH-dependent catalytic activities, such as reduction of organic hydroperoxides and isomerization of several unsaturated compounds, as well as non-catalytic functions related to the capture of carcinogens, intracellular transport of hydrophobic ligands, and modulation of signal transduction pathway [129]. Since xenobiotic metabolizing enzymes constitute an important line of defense against a variety of carcinogens [75], inherited differences in the capacity of these enzymes might be an important genetic factor leading to susceptibility to cancer [80].
The GSTs Mu-1 and Theta-1 are different isoforms belonging to two of the eight families or classes of the mammalian cytosolic or soluble GSTs [129,132], which appear to be primarily involved in the metabolism of foreign chemicals (xenobiotics), such as carcinogens, environmental pollutants and cancer chemotherapeutics, as well as the detoxication of potentially harmful endogenous reactive compounds (products of oxidative stress) [129]. In humans, a significant number of genetic polymorphisms among the soluble GSTs have been described [129,133]. Among them are the deletion polymorphisms (deletion of the whole gene) resulting in the lack of active enzyme in the GSTM1 (locus 1p13.3) and GSTT1 (locus 22q11.2) genes, which encode respectively the GSTs Mu-1 and Theta-1 enzymes [129,131,133–136]. Although variation in GST alleles is very common in populations worldwide and will presumably make a significant contribution to individual differences (intra and inter-ethnic) [128,129], homozygous deletions in the GSTM1 or GSTT1 results in no detectable enzyme activity, and may diminish the ability to detoxify various carcinogens [24,128,131], predisposing to disease and responsiveness to therapeutic agents [133]. Therefore, occupational and environmental exposures to xenobiotics or increased oxidative stress, as well as certain hobbies and/ or lifestyle exposures may confer augmented susceptibility to CML in those individuals carrying one or both these deletions in homozygosis.
Association studies involving the deletion polymorphisms in GSTM1 and GSTT1 genes with risk of CML have been carried out in different populations. However, most of them just analyzed this risk for the null genotypes (odds ratio with 95% confidence intervals), where some results of positive association with CML risk were observed only for the GSTT1 null genotype, as shown in table 3.
| Authors, year | Ethnicity | Polymorphisms | Characteristics of cases | Characteristics of controls | Polymorphisms | Case | Control | OR (95% CI) | P value |
| Lemos et al., 1999 [75] | White | GSTM1 | 11 patients with CML, from a cohort of 160 cases with hematologic neoplasias (in cohort, 53% males, mean age 49.9 21.1 yrs) | 128 healthy controls with no history of cancer or other chronic diseases, 56% females, mean age 30.8 14.2 yrs | GSTM1 | ||||
| Present | 2(18.2) | 54(42.2) | – | – | |||||
| Null | 9(81.8) | 74(57.8) | – | NS | |||||
| Löffler et al., 2001[76] | White | GSTM1, GSTT1 | 141 patients, 55% males | 150 healthy controls, 66% males | GSTM1 | ||||
| Present | 64(45.4) | 66(43.2) | – | – | |||||
| Null | 77 (54.6) | 84 (56.8) | 0.92 (0.58-1.46) | 0.71 | |||||
| GSTT1 | |||||||||
| Present | 110(88) | 124(82.7) | – | – | |||||
| Null | 31 (22.0) | 26 (17.3) | 1.34 (0.75-2.40) | 0.32 | |||||
| Lourenço et al., 2005 [77] | Mixed | GSTM1, GSTT1 | 125 patients (86% white), 58% males, mean age 39.0 16.4 yrs |
341 controls, 58% males, mean age 53.0 4.3 yrs, ethnicity matched | GSTM1 | ||||
| Present | 71(56.8) | 192(56.3) | – | – | |||||
| Null | 54(43.2) | 149(43.7) | 0.98 (0.65-1.50) | 1 | |||||
| GSTT1 | |||||||||
| Present | 102(81.6) | 281(82.4) | – | – | |||||
| Null | 23(18.4) | 60(17.6) | 1.06 (0.62-1.80) | 0.95 | |||||
| Mondal et al., 2005 [78] | Indian | GSTM1, GSTT1 | 81 patients, 70% males, median age 40 (3–81) yrs | 123 healthy controls without history of cancer or other chronic diseases, 55% males, median age 35 (19–65) yrs, geographically and ethnicity matched | GSTM1 | ||||
| Present | 58 (71.6) | 89 (72.3) | 1 | – | |||||
| Null | 23 (28.4) | 34 (27.7) | 1.04 (0.55-1.93) | 0.91 | |||||
| GSTT1 | |||||||||
| Present | 65 (80.2) | 114 (92.7) | – | – | |||||
| Null | 16 (19.8) | 9 (7.3) | 3.12 (1.3-7.45) | 0.008 | |||||
| Hishida et al., 2005 [79] | East Asian | GSTM1, GSTT1 | 51 patients, 63% males, mean age 47.4 (21–78) yrs | 476 controls, 61% males, mean age 49.7 (17–89) yrs | GSTM1 | ||||
| Present | 25 (49.0) | 227 (47.7) | – | – | |||||
| Null | 26 (51.0) | 249 (52.3) | 0.95 (0.53-1.69) | 0.857 | |||||
| GSTT1 | |||||||||
| Present | 22 (43.1) | 238 (50.0) | – | – | |||||
| Null | 29 (56.9) | 238 (50.0) | 1.32 (0.74-2.36) | 0.353 | |||||
| Bajpai et al., 2007 [80] | Indian | GSTM1, GSTT1 | 80 patients, 73% males, mean age 36.2 10.9 yrs | 105 healthy controls, 56% males, mean age 36.8 11.3 yrs, and ethnicity matched | GSTM1 | ||||
| Present | 56 (70%) | 79 (75.2%) | – | – | |||||
| Null | 24 (30%) | 26 (24.7%) | 1.30 (0.65-2.63) | 0.530 | |||||
| GSTT1 | |||||||||
| Present | 64 (80%) | 96 (91.4%) | – | – | |||||
| 2.67 (1.03-7.01) | 0.0417 | ||||||||
| Chen et al., 2008 [81] | East Asian | Null GSTM1 Present Null GSTT1 Present |
16 (20%) 50 (46.3) 58 (53.7) 57 (52.8) |
9 (8.5%) 91 (44.6) 113 (55.4) 104 (51.0) |
|||||
| 1 | – | ||||||||
| 0.95 (0.59-1.53) | NS | ||||||||
| 1 | – | ||||||||
| 0.91 (0.57-1.45) | NS | ||||||||
| Taspinar et al., 2008 [82] | Turkish | Null GSTM1 Present |
100 (49.0) 59 (55,1) |
51 (47.2) 75 (57,7) |
|||||
| 1 | – | ||||||||
| Null | 48 (44,9) | 55 (42,3) | 1.11 (0.66-1.86) | 0.693 | |||||
| GSTT1 | |||||||||
| Present | 64 (59,8) | 105 (80,8) | 1 | – | |||||
| Null | 43 (40,2) | 25 (19,2) | 2.82 (1.58-5.05) | < 0.001 | |||||
| Souza et al., 2008 [83] | Mixed | GSTM1, GSTT1 | 53 patients, 57% males, mean age 41.0 21 yrs | 304 healthy controls, 57% females, mean age 53.0 4.3 yrs | GSTM1 | ||||
| Present | 36(67.9) | 190(62.5) | – | – | |||||
| Null | 17(32.1) | 114(37.5) | – | NS | |||||
| GSTT1 | |||||||||
| Present | 43(81.1) | 265(87.2) | – | – | |||||
| Null | 10(18.9) | 39(12.8) | – | NS | |||||
| Ovsepyan et al., 2010 [84] | White/Russian | GSTM1, GSTT1 | 83 patients, mean age 56.9 yrs | Control group included 205 healthy unrelated volunteers. The average age of the control patients did not significantly differ from that in the CML patient group. |
GSTM1 | ||||
| Present | 39 (46.99) | 111 (54.15) | 0.75 (0.45-1.25) | 0.33 | |||||
| Null | 44 (53.01) | 94 (45.85) | 1.33 (0.80-2.22) | 0.33 | |||||
| GSTT1 | |||||||||
| Present | 63 (75.90) | 178 (86.83) | 0.48 (0.25-0.91) | 0.04 | |||||
| Null | 20 (24.10) | 27 (13.17) | 2.09 (1.10-3.99) | 0.04 | |||||
| Mahmoud et al., 2010 [85] | Egyptian | GSTM1, GSTT1 | 30 patients, mean age 41.7 yrs | 20 age and sex matched healthy controls. | GSTM1 | ||||
| Present | 16(53.3) | 12(60) | – | – | |||||
| Null | 14(46.7)) | 8(40) | 1.313 (0.42-4.13) | 0.0642 | |||||
| GSTT1 | |||||||||
| Present | 12(40) | 17(85) | – | – | |||||
| Null | 18(60) | 3(15) | 8.50 (2.04-35.46) | 0.002 | |||||
| Lordelo, 2011 [74]; Lordelo et al., 2012 [24] |
Mixed | GSTM1, GSTT1 | 105 CML patients | The control group consisted of 273 non-related healthy volunteers of both genders. | GSTM1 | ||||
| Present | 50 (47.6) | 97 (35.5) | 1.65 (1.05-2.60) | 0.031 | |||||
| Null | 55 (52.4) | 176 (64.5) | 0.61 (0.38-0.96) | 0.031 | |||||
| GSTT1 | |||||||||
| Present | 84 (80) | 208 (76.2) | 1.23 (0.70-2.13) | 0.473 | |||||
| Null | 21 (20) | 65 (23.8) | 0.82 (0.47-1.42) | 0.473 | |||||
| Özten et al., 2012 [86] | White / Turkish | GSTM1, GSTT1 | 106 patients, 56.6% males, mean age 35.1 yrs | The control group consisted of 190 healthy unrelated volunteers without history of cancer or other chronic diseases 56.3% males, mean age 38.3 yrs | GSTM1 | ||||
| Present | 58 (54.7) | 109 (57.4) | 1 | – | |||||
| Null | 48 (45.3) | 81 (42.6) | 1.11 (0.69, 1.80) | 0.714 | |||||
| GSTT1 | |||||||||
| Present | 59 (55.7) | 155 (81.6) | 1 | – | |||||
| Null | 47 (44.3) | 35 (18.4) | 3.53 (2.08, 6.00) | < 0.0001 | |||||
| Bhat et al., 2012 [87] | Indian | GSTM1, GSTT1 | 75 CML patients (43 males and 32 females; age (mean ± S.D) 42.3 ± 13.4 years) | 124 unrelated non-malignant controls (76 male and 48 females; age (mean ± S.D) 41.5 ± 12.9) | GSTM1 | ||||
| Present | 44 (59%) | 81(65%) | 1 | – | |||||
| Null | 31 (41%) | 43 (35%) | 1.32 (0.73 - 2.40) | 0.4295 | |||||
| GSTT1 | |||||||||
| Present | 48 (64%) | 98 (79%) | 1 | – | |||||
| Null | 27 (36%) | 26 (21%) | 2.12 (1.12 - 4.02) | 0.0308 |
Table 3: Association studies among the deletion polymorphisms in the glutathione S-transferases GSTM1 and GSTT1 in CML patients and controls.
However, one of the two articles that analyzed both genotypes of the deletion polymorphisms (null and non-null genotypes) in GSTM1 and GSTT1 genes found a positive association between CML risk with GSTM1 non-null genotype (OR = 1.649; 95% CI = 1.05-2.6), while GSTM1 null genotype decreased this risk (OR = 0.606; 95% CI = 0.21- 0.77) [24]. Although it is unbelievable that the presence of an enzyme, which operates in carcinogen detoxification, could increase CML risk, in several instances GST activity does not result in the detoxification of xenobiotics and may even lead to a more reactive compound than the parental one. Additionally, incomplete detoxification by GST may occur with certain esters, ethers, and organic phosphates, and the unconjugated cleavage product still provides a chemical threat to the cell. Moreover, while toxification by GST is undesirable in normal circumstances, it can be exploited in cancer chemotherapy to treat tumors that overexpress GST [127]. Thus, these effects need to be further investigated. Also, the GSTM1 gene demonstrates polymorphisms that arise from homo and heterozygotic combinations of the GSTM1*0, GSTM1*A and GSTM1*B alleles [133,136], and although it is believed that GSTM1*A and GSTM1*B alleles encode proteins that are catalytically identical [130,133], a linkage disequilibrium between GSTM1 and GSTM3 (both at locus 1p13.3) has been reported, and this may alter the expression of these class Mu transferases. For example, it has been demonstrated that subjects with GSTM1*A/GSTM3*B should express GSTM3 at higher levels than those with GSTM1*0/GSTM3*A or GSTM1*B/GSTM3*A [133], and this needs to better investigated, at least for those subjects that are less responsive or non-responsive to therapeutic agents.
Still considering possible genetic interactions, for the most part, polymorphisms in individual GST genes do not confer a markedly increased risk of cancer, but combinations of the GST M1 and T1 alleles taken together or with alleles of other genes encoding for detoxification or antioxidant enzymes are likely to have an additive effect in conferring predisposition to cancer or to influence responsiveness to chemotherapeutic agents in its treatment [129]. Thus, some of the association studies showed in table 3 also evaluated interactions among the deletion polymorphisms in the GSTM1 and GSTT1 genes with other gene polymorphisms related to xenobiotic metabolizing enzyme of phases I (eg. cytochrome P450s – CYPs or P450s) and/or II (eg. N-acetyltransferase 2 - NAT2; NAD(P)H:quinone oxidoreductase 1 - NQO1) in the CML risk [75,76,79,81–83,86]. However, results of association with increased CML risk were only found for the interactions between GSTM1normal/GSTT1null and/or GSTM1null/GSTT1null genotypes [82,83,86], although it has been proposed that this risk would be modulated little by GSTT1 and GSTM1 deletions [79]. These results suggested an interaction between GSTT1 and GSTM1 genotypes and that GSTT1 null genotype could be the significant risk factor for CML [82]. Nevertheless, as previously mentioned, associations between GSTT1 null genotype and increased CML risk were not found in many studies [24,77,79,81]. Besides the small sample size of some studies resulting in very broad confidence intervals, such contrary findings may also reflect geographical differences in the type of environmental carcinogens to which different populations are exposed [75]. They also may reflect differences in CML risk among the different ethnic groups, since inter- and intra-ethnic differences in the allele frequencies of GSTM1 and GSTT1 null genotypes have been documented worldwide [128]. Thus, studies dealing with a large population in several ethnicities should be encouraged to better understand the biological significance of these polymorphisms in CML susceptibility in each ethnic group, as well as in understanding their role in the variability of the disease course and in responsiveness to therapeutic agents, mainly for women <50 years, where analysis of survival carried out from the SEER database suggested possible higher rates of TKI resistance for the African-American group [10].
Although BCR-ABL transcripts are known to be associated uniquely with acute and chronic leukemia, studies using sensitive techniques of reverse transcriptase-polymerase chain reaction (RTPCR) have detected small amounts of such transcripts in leukocytes of normal healthy adults [137,138] and children [137], as well as in human hematopoietic non-CML cell lines [138]. This suggests that such forms of illegitimate genetic recombination may occur regularly in hematopoietic precursors and in cultured cell lines as a consequence of an inherent basal level of genomic instability [138]. However, only infrequently do the cells acquire the capacity to produce leukemia in humans. Although success in producing a leukemic phenotype depends on the fusion gene structure producing a functional protein and also the occurrence of chromosomal translocation in a relatively early precursor cell with self-renewal capacity, which were not detected in several normal leukocytes and in the non-CML cell lines [138], it is also possible that these transcripts represent “errors” that occur because of the close proximity of BCR and ABL1 genes during the late S-phase (S to G2 transition) of the cell cycle, and that immune surveillance prevents the emergence of CML in individuals who remain healthy [20,139]. Therefore, the possible association of leukemia with haptoglobin (Hp), an acute-phase plasma α2-glycoprotein with several functional properties, including antioxidant, anti-inflammatory and immunomodulatory ones, was first suggested by Latner and Zaki [88,93,140].
The main biological function of Hp is binding free hemoglobin, removing it from the circulation and limiting hemolysis and iron loss during normal erythrocyte turnover, thereby preventing oxidative damage mediated by excess iron in the circulation. As a result, Hp functions as an antioxidant [98,141,142]. In addition, Hp has the ability to regulate immune cell responses and host immunity, modulating the balance of helper T-cell types 1 and 2 (Th1/Th2) within the body, and thus supporting proliferation and functional differentiation of B and T cells as part of homeostasis and in response to antigen stimulation [141–143]. Because of its immunomodulatory property, Hp may inhibit or stimulate the immune response, and its concentration in malignancies and in inflammatory and infectious processes is elevated [142]. However, Hp of humans is polymorphic, and its serum levels are phenotype-dependent [144,145], as is its ability to block hemoglobininduced oxidative stress and damage and its anti-inflammatory activity [24,141,143,145-147]. Thus, many clinical studies have demonstrated a link between Hp polymorphism and a broad range of pathological conditions, often with divergent clinical consequences, and such associations probably reflect functional differences among the phenotypes [141,145-148].
The human Hp gene is located on chromosome 16 (locus 16q22.1) and its molecular variation was described by Smithies [149] , who identified by starch gel electrophoresis three major phenotypes, Hp1-1, Hp2-1, and Hp2-2, which are controlled by two autosomal codominant alleles, Hp*1 and Hp*2. After that, on starch gels with urea, the Hp*1 allele revealed two subtypes, Hp*1S and Hp*1F, and then the three common types of Hp could be subdivided into a total of six, which are products of expression of the combination of the alleles Hp*1F, Hp*1S and Hp*2 [142,145,147,149,150].
In view of the above explanations and also that: (1) Hp binds to different immunologic cells by specific receptors, and among them are the natural killer (NK) cells [141], an important part of immune surveillance [151]; (2) the three main Hp phenotypes have the same binding affinities for hemoglobin [143], but Hp2-2 removes iron to the extravascular space more slowly because it is a larger molecule [133]; so, the antioxidant capacity of the Hp 1-1 phenotype is higher than Hp 2-2 because free hemoglobin remains in the circulation longer and causes greater oxidative stress [142,144,152]; (3) ALL, AML, and CML have been associated with an increased incidence of Hp 1-1, but the explanation of this phenomenon is difficult [93]; and finally (4) genome-wide analysis of gene-expression profiles in CML cells using a cDNA microarray has shown haptoglobin (HP1) to be one of the upregulated elements [153]; these apparently divergent data can only be understood through additional characterization of the Hp*1 subtype polymorphisms (Hp*1S and Hp*1F). However, only one study carried by Lordelo et al. which worked with a mixed population (Brazilian), researched a possible association of CML with these Hp*1 subtype polymorphisms [24] in a case control study, as can be seen in table 4. Even though no association was found in this work between a particular Hp genotype when considering the whole population, significant results were obtained for individuals of black skin color, where Hp1F- 1S individuals presented an increased risk (OR = 7.200; 95% CI = 0.94- 54.94; p = 0.037), while Hp1S-2 individuals, a decreased risk (OR = 0.619; 95% CI = 0.44-0.87; p = 0.011) [24].
| Authors, year | Ethnicity | Polymorphisms | Characteristics of cases | Characteristics of controls | Types/ Subtypes | Case N(%) |
Control (N/%) | OR (95% CI) | P value |
| Latner and Zaki, 1960 [88] | White (England) | Haptoglobin | 10 adult patients with CML | 30 normal individuals | 1-1 | 4(40) | 3(10) | – | – |
| 2-1 | 4(40) | 15(50) | – | – | |||||
| 2-2 | 2(20) | 12(40) | – | – | |||||
| Peacock, 1966 [89] | White | Haptoglobin | 24 adult patients with CML | 101 patients with a variety of disease, not including leukemia. | 1-1 | 4(16.7) | 15(14.8) | – | – |
| 2-1 | 6 (25) | 33(32.7) | – | – | |||||
| 2-2 | 14(58.3) | 53(52.5) | – | – | |||||
| *Baxi and Camoens, 1969 [90]; Blake et al., 1971 [91]; Naik et al., 1979 [92] |
India | Haptoglobin | 85 adult patients with CML | 1205 normal controls for the study by Naik et al., 1979, which were not provided by authors. Blake et al. 1971, Hp types in nine population groups from India. Baxi and Camoens, 1969, normal control from north and western India | 1-1 | 5(5.9) | 36(2.98) | – | – |
| 2-1 | 23(27.05) | 458(38) | – | – | |||||
| 2-2 | 57(67.05) | 1025(85) | – | – | |||||
| *Fröhlander, 1984 [94] | Sweden | Haptoglobin | 22 adult patients with CML | 2297 normal controls | 1-1 | 4(18.2) | 311(13.5) | – | – |
| 2-1 | 8(36.4) | 1091(47.5) | – | – | |||||
| 2-2 | 10(45.4) | 895(39) | – | – | |||||
| Michell et al., 1988 [95] | Haptoglobin | 22 adult patients with CML | 361 blood donors | 1-1 | 5(22.7) | 56(15.5) | – | – | |
| 2-1 | 10(45.5) | 175(48.5) | – | – | |||||
| 2-2 | 6(27.3) | 124(34.3) | – | – | |||||
| Campregher et al., 2004 [96] | Mixed | Haptoglobin | 78 adult patients with CML | 210 blood donors from the same geographical region | 1-1 | 13(16.7) | 45(22.9) | – | – |
| 2-1 | 42(53.8) | 108(54.8) | – | – | |||||
| 2-2 | 23(29.5) | 44(22.3) | – | – | |||||
| Lordelo, 2011 [74]; Lordelo et al., 2012 [24] |
Mixed | Haptoglobin | 105 CML patients | 273 non-related healthy volunteers of both genders | 1F-1F | 4(3.81) | 16(5.9) | 0.636 (0.208-1.949) | 0.425 |
| 1F-1S | 12(11.43) | 18(6.6) | 1.828 (0.848-3.940) | 0.119 | |||||
| 1S-1S | 5(4.76) | 28(10.3) | 0.438 (0.164-1.165) | 0.090 | |||||
| 1F-2 | 13(12.38) | 36(13.2) | 0.930 (0.472-1.833) | 0.835 | |||||
| 1S-2 | 32(30.48) | 93(34.1) | 0.848 (0.522-1.378) | 0.506 | |||||
| 2-2 | 39(37.14) | 82(30) | 1.376 (0.858-2.209) | 0.185 |
Table 4: Association studies among haptoglobin types and subtypes in CML patients and controls.
While Hp*1 and Hp*2 alleles have always been found in every population examined to date, there is a significant difference in their distribution in populations worldwide, as well as in the distribution of the Hp*1F and Hp*1S allele frequencies [146,147], which suggests that particular populations are susceptible to particular diseases [146]. Moreover, even though the Hp*1F allele is rarer than the Hp*1S allele, the higher frequencies of the Hp*1 allele in South America and Africa seems to be linked to a higher Hp*1F frequency in studies that make such data available [146,147].
Thus, to avoid spurious associations and especially loss of some important association, Hp*1 subtypes cannot be treated as a single block of Hp 1-1 phenotypes in association studies, particularly those involving South America, Africa or admixed populations. This has already been indicated in other studies carried out with Brazilian athletes, which verified differences in the biological responses among Hp*1 alleles, particularly those involving oxidative stress [154,155]. Therefore, studies evaluating a possible association between Hp subtypes and those leukemia associated with an increased incidence of Hp 1-1 (CML, ALL and AML) should be encouraged in several ethnicities, particularly in South America and Africa, where Hp*1F allele frequency is higher. These studies are particularly important after the findings of Mandal et al. [10], where, as previously mentioned, the survival rates of African-American men <50 years with CML was significantly lower compared to those of Caucasians in the pre-TKI era, while the same age group of African-American women had lower relative survival rates also compared to Caucasian women in the post- TKI era [10].
Since little is known about individual susceptibility to CML, and the mechanisms behind disease progression are not fully understood, the MTHFR, GSTM1, GSTT1 and Hp polymorphisms are good candidates for a possible genetic susceptibility to the induction of the Ph chromosome and the emergence of CML. However, since these polymorphisms vary among ethnic groups, and populations worldwide vary considerably in their predisposition to diseases and in the allele frequencies of pharmacogenetically important loci, studies in several ethnicities are required to better understand the biological significance of these polymorphisms in CML susceptibility, as well as in understanding the variability of the disease course and the patients’ response to medication. As it has been demonstrated that there are differences in the incidence and death rates of CML by gender and race/ ethnicity pre- and post-TKI, and various association studies focusing the addressed polymorphisms analyzed relatively small groups of patients and controls which resulted in very broad intervals of statistical confidence of relative risks, studies with large populations worldwide are required to remedy these methodological problems and to confirm the role of MTHFR, GSTM1, GSTT1 and Hp polymorphisms in CML.