Journal of Chromatography & Separation Techniques
Open Access
ISSN: 2157-7064
ISSN: 2157-7064
Research Article - (2016) Volume 7, Issue 5
A high performance liquid chromatographic method characterized by its rapidness and sensitivity was developed and validated for quantitation of Cilostazol (CIL) and Telmisartan (TEL) in raw material, their synthetic mixture using isocratic technique and monolithic C8 column (3 mm × 4.6 mm i.d., 2 μm pore size highly porous). The mobile phase composed of acetonitrile:0.03 M dihydrogen phosphate buffer (40:60, v/v) at pH 4.5. Quantification was achieved through UV detection at 257 nm using flow rate of 1 mL/min, and Dipyridamole (DIP) was used as internal standard. DIP, CIL, and TEL, retention times were 2.2, 3.9 and 5.1 min. respectively. Peak area ratios of each drug to Dipyridamol internal standard were plotted against concentration of each drug and linear relations were obtained in the range of 0.5-15 μg/mL for CIL 0.25-20 μg /mL for TEL. The method was successfully utilized for the assay of CIL and TEL synthetic mixture. Stability-indicating assay methods (SIAM) were mentioned for separation of CIL in presence of its degradation products. Cilostazol was subjected to acid and alkali hydrolysis, oxidation and photochemical degradation. It was stable under acidic, basic and ultraviolet degradation conditions, but undergoes oxidative degradation, therefore the drug was separated from its oxidative degradation product using our proposed high performance liquid chromatographic and derivative ultraviolet
spectrophotometric
methods. The first derivative method (D1) depend on measuring the amplitude values at 227 and 257 nm for Cilostazol and oxidative degradate, respectively. From calibration plots, linearity was obtained in the range of 1-35 μg/mL for Cilostazol and 2-50 μg/mL for oxidative degradate. Chromatographic separation of Cilostazol from its oxidative degradate was proceeded using the same mobile phase at pH 3.3. All methods were validated statistically as per International Conference on Harmonization (ICH) recommendations for the studied drugs and Cilostazol oxidative degradate in the concentration range of the suggested methods se ion-pair liquid chromatography was described.
Keywords: Cilostazol; Telmisartan; High performance liquid chromatography; Stability-indicating assay method (SIAM); Oxidative degradation; First derivative spectrophotometric method
Cilostazol (CIL) (Figure 1a) 6-[4-(1-Cyclohexyl–1H-tetrazol–5–yl) butoxyl]-3,4–dihydro–2(1H)-quinolinone [1], is a phosphodiesterase inhibitor with anticoagulant and vasodilating behavior applied for peripheral vascular disease treatment and for reduction of symptoms of intermittent claudication [2]. Telmisartan (TEL) (Figure 1b) 4′-[(1,4′-Dimethyl–2′-propyl[2,6′-bi–1H-benzimidazol]- 1′-yl) methyl]-[1,1′biphenyl]-2–carboxylic acid [1], is an angiotensin receptor blocker used for management of hypertension [2]. CIL and TEL are official in USP [3], TEL is official in BP [4]. Literature survey yielded some published methods for CIL assay include: Spectrophotometry as single drug [5-8], as mixture with other drugs [9-11], HPLC methods as single drug [12-15], and with other drugs [16,17]. Several analytical methods were reported for Telmisartan assay include: Spectrophotometry as single drug [18-20], as mixture with other drugs [21-27], Spectrofluorimetric methods as a mixture with other drugs [28,29]. Also many HPLC procedures have been published for its quantitation in its single form [30-33] and as mixture with others [34-40]. Therefore, it was desirable to develop simple, accurate, cheap and rapid procedure that could be utilized for simultaneous assessment of Cilostazol and telmisartan. It is not the first time for simultaneous HPLC determination of CIL and TEL, since there was a previously reported method [17], but our proposed method was superior in that it possess the advantages of high sensitivity over the reported one, LOQ of 0.11 and 0.2 ug/ml for CIL and TEL respectively and with smaller analysis time of 5 min than the reported one (10 min). Also, our proposed HPLC method was extended for determination of both drugs in their single tablet formulation. Moreover, stability study was not recomended in the reported method, but our proposed method could be used for determination of CIL in presence of its oxidative degradation. The developed proposed method presents an economic, simple, sensitive procedure for simultaneous assay of cilostazol and telmisartan in their synthetic mixture and also used for separation of cilostazol from its oxidative product of degradation as an application to stability testing. First derivative spectrophotometric method was used for quantitation of cilostazolin presence of its oxidative degradate. Validation of the obtained results as per ICH guidelines showed that the proposed methods are of high accuracy and good precision.

Figure 1a: Structural formula of Cilostazol.

Figure 1b: Structural formula of Telmisartan.
Apparatus
Perkin Elmer TM Series 200 Chromatograph joined with a Rheodyne injector valve with a 20 μL loop and UV/VIS detector was utilized for HPLC assay. Chromatograms were recorded on a PC attached to the device. Solvent degasser was used for mobile phases degassing. Spectrophotometric analysis was accomplished using a Shimadzu (Kyoto, Japan) UV-1601 PC, UV-Visible double-beam spectrophotometer with matched 1 cm path-length quartz cells. The spectra of the studied drugs were measured in 1 cm quartz cells and recorded on a fast scan speed, setting slit width to be 1 nm against solvent blank using wavelength range (200-400) nm using Δλ=8 nm and scaling factor=10 to obtain smoothly spectra. A Consort NV P-901 pH Meter (Belgium) was utilized for pH adjustment.
Materials and reagents
All used chemicals and reagents must be of high analytical grade and solvents used must be HPLC grade. Cilostazol, pure sample (Batch # 061500080) was kindly provided by European Egyptian Pharmaceuticals Company, Cairo, Egypt. Its purity was determined by official method (3)100.18%. Telmisartan was kindly supplied by biopharma Pharmaceutical Company, Cairo, Egypt. Its purity was determined by official method (3)100.02%. Laboratory prepared mixture consists of 40 mg CIL, 10 mg TEL, 20 mg talc powder, 15 mg maize starch, 15 mg lactose and 7 mg magnesium stearate Dipyridamol was kindly provided by Mepaco Pharmaceutical Corporation, Cairo, Egypt, with purity of 100.05%. Claudol tablet (100 mg of cilostazol) was manufactured by Sabaa International Company for Pharmaceuticals and Chemical Industries S.A.E. (M.O.H. Reg. No: 2440/2006) was obtained from commercial sources in local pharmacy. Micardis® tablet labeled to contain 40 mg of telmisartan, product of Boehringer Ingelheimpharma GmbH & Co. HG (Reg. No: 303028-01) was purchased from commercial sources in the local market. Acetonitrile (HPLC grade) was obtained from Sigma-Aldrich (Germany). -Orthophosphoric acid (85% w/v) was supplied from Riedel-deHäen (Germany). Sodium hydroxide and sodium dihydrogen phosphate were purchased from Adwic Co. (Cairo, Egypt).
Chromatographic conditions
The chromatographic assay was done using monolithic C8 column (3 mm × 4.6 mm i.d., 2 μm pore size highly porous column), A mixture of acetonitrile and 0.03 M dihydrogen phosphate buffer (40:60, v/v) was the selected mobile phase for elution, the pH was adjusted to 4.5 with 0.02 M orthophosphoric acid and filtered through a 0.45 μm membrane filter (Millipore, Ireland) and pumped at a flow rate of 1 mL/min. Such experiment was performed at room temperature and the UV detection was carried out at 257 nm. Dipyridamol was picked as the most appropriate internal standard
Stock solutions preparation
Standard stock solution of 100.0 μg/mL was used for both drugs and internal standard, using methanol as dissolving solvent for the three drugs. By suitable dilution of the stock solutions using the mobile phase, working solutions were obtained. Preserving the solutions in the refrigerator make them stable for at least a month without alteration.
Assay of cilostazol and telmisartan in raw materials
Construction of calibration plots: Cilostazol and Telmisartan working solutions were prepared from the previously mentioned stock solutions by serial dilution using the eluent to obtain final concentrations of (0.5-15) μg/mL for CIL and (0.25-20 μg/mL) for TEL into a series of 10 mL measuring flasks. 3 μg/mL of Dipyridamol stock solution was added to each flask as internal standard. Then, the final volume was adjusted using the mobile phase (pH=4.5) to the mark and mixed well. 20 μL aliquots were injected (triplicate) using the mobile phase as the eluent under the optimized chromatographic parameters. The ratios of peak area (drug/I.S.) against the final concentration of each drug in μg/ml were plotted. Alternatively, the corresponding regression equations were established.
Analysis of CIL/TEL in their synthetic mixtures: Aliquots of standard solutions of CIL and TEL keeping the curative ratio of 4:1, respectively were Transferred into a series of 10.0 mL volumetric flasks, diluted using mobile phase and mixed well then analyzed using the procedure stated under "Construction of calibration plots". From the obtained calibration plots, the percentage recoveries were calculated. Also percentage recoveries can be obtained from the corresponding regression equations.
Assay of the studied drugs in single tablet formulations: Ten Claudol tablets were exactly weighed, finely pulverized and mixed well, and then quantity of the powder equivalent to 100 mg of CIL was transferred into a 100.0 mL volumetric flask and about 70.0 mL of methanol were added. The flask contents were subjected to sonication for 30 min, completed with methanol to the final volume and filtered. Working solutions were prepared using the same solvent for dilution and analyzed using the general procedure as illustrated under “construction of calibration graph”. Simultaneously, Ten Micardis tablets were accurately weighed, finely grinded and thoroughly mixed, then a quantity of the resulted powder equivalent to 100 mg of TEL was transferred into a 100.0 mL volumetric flask and continue the procedure as described in case of Claudol tablet. From the previously plotted calibration graph or by employing the corresponding regression equation, the nominal content of the tablets can be calculated.
Procedure for Cilostazol degradation products preparation: One mL aliquots of CIL standard stock solution (10 mg/mL) were transferred into a series of 25 mL volumetric flask to reach final concentration equal 400.0 μg/mL, and then 5 ml of 2M hydrochloric acid, 2M sodium hydroxide, or 15% hydrogen peroxide was added to prepare acidic, alkaline and oxidative degradation product respectively. The solutions (acidic and alkaline degradation) were refluxed at 100°C for 6 hrs, left in boiling water bath for 30 minutes at 80°C for oxidative degradation. In case of the UV degradation, the methanolic solution of CIL was subjected to Deuterium lamp at 254 nm in a wooden cabinet distance of 15 cm for 9 hours. Blank experiment was done simultaneously. After cooling the solutions were neutralized with 2M sodium hydroxide or 2M hydrochloric acid for acidic and alkaline degradation respectively, and completed with the mobile phase to the final volume. And then apply the proposed HPLC method for separation. It was found that no peaks were separated in case of acidic, alkaline and photochemical degradation and the peak area of CIL was not affected so, the drug is stable against such mentioned conditions. In case of oxidative degradation, noticeable increase in Cilostazol peak area was occurred. On changing the pH to 3.3 oxidative degradates was separated from Cilostazol. Also, derivative UV spectrophotometric method was established for quantitations of CIL with existence of its oxidative degradate.
Separation of cilostazol in presence of its oxidative degradation product using the suggested HPLC method
Cilostazol and its oxidative degradate can be separated with good resolution using our proposed HPLC method at pH 3.3. Such modification in pH is due to the great overlap between cilostazol and its oxidative degradate at pH 4.5. The retention times are 3.67 and 4.39 min. for oxidative degradate and cilostazol simultaneously. Cilostazol can be assayed in presence of different percentages of oxidative degradate.
First derivative spectrophotometric method for quantitation of cilostazol with existence of its oxidative degradation product
Aliquots of CIL and oxidative degradate standard solutions covering the concentration range of 1.0-35.0 μg/mL and 2.0-50.0 μg/mL, simultaneously were transferred into two series of 10 mL volumetric flasks and the solutions were completed with methanol to the final volume and mixed well. The zero-order absorption spectra of CIL and its oxidative degradate were scanned against methanol showing great overlap. The first derivative spectra (1D) of cilostazol and its oxidative degradate were scanned in the wavelength range (200- 350) nm using Δλ=8 nm with zero crossing point recorded at 228 and 257 nm, simultaneously. The peak amplitude of the first derivative experiment was plotted against final concentration (μg/mL) to get the calibration graphs. Alternatively, the corresponding regression equations were derived (Figure 2).

Figure 2: Zero order absorption spectra overlay of 10 μg/mL CIL (a) and 10 μg/mL TEL (b) in methanol.
CIL and TEL were separated using accurate and selective HPLC method and show well-resolved symmetrical peaks with good resolution in a short time nearly 5 min. using the optimized chromatographic conditions. Figure 3 shows typical chromatogram of a synthetic mixture of both drugs at their curative pharmaceutical ratio (4:1) using the proposed HPLC procedure. It is also permitted the quantitation of both drugs separately in their tablets (Figure 4A and 4B). Also, HPLC method was used for selective determination cilostazol in prescence of its oxidative degradate with satiafactory results at pH 3.3 (Figure 5). The drug was found to be completely degraded, and it was confirmed by injecting Cilostazol (10 μg/mL) and oxidative degradate (any concentration) on a separate runs then injecting a mixture of Cilostazol (10 μg/mL) and oxidative degradate (any concentration). It was regarded that peak area of CIL was the same in both cases. For spectrophotometric method of assay the UV spectrum of CIL solution in methanol showed absorption maxima at 220, 260, while that of oxidave degradate showed absorption maxima at 270 nm (Figure 6). So, great overlap between absorption spectra of CIL and degradation product in Zero order that prevent the direct quantitation of CIL in presence of its oxidative degradation product, therefore the first derivative was carried out for the quantification of CIL in the presence of its degradation product. Figure 7a and 7b shows the first derivative spectra (D1) of CIL and its oxidative product and shows zero crossing point for each. The effect of different Δλ [2,4,6,8] on the developed first derivative spectra was studied, showing that Δλ=8 was the selected one for best determination of both analytes. Using Δλ=8, CIL was quantified in presence of its oxidative degradate at zero crossing point (228) and quantitation of the degradation product in presence of CIL at zero crossing point (275). Figure 8 shows first derivative absorption spectra of CIL and its oxidative degradate in different synthetic ratios.

Figure 3: Typical chromatogram of synthetic mixtures under the described chromatographic conditions; acetonitril: phosphate buffer (40:60, v/v) at pH 4.5 and a flow rate of 1 mL/min. where: A: Solvent front; B: Dipyridamol (20 μg/mL); C: Cilostazol (12 μg/mL); D: Telmisartan (3 μg/mL)

Figure 4: 4A: Typical chromatogram of the Cilostazol determination in Claudol tablet where: A: Solvent front B: Dipyridamol I.S (3.0μg/mL) C: Cilostazol (10μg/mL). 4B: Typical chromatogram of the Telmisartan determination in Micardis tablet where: A: Solvent front B: Dipyridamol I.S (3.0μg/mL) C: Telmisartan (10μg/mL).

Figure 5: Typical chromatogram of cilostazol in presence of 100% of its oxidative degradation product (10 μg/mL of each) under the described chromatographic conditions; acetonitrile: phosphate buffer (40:60, v/v) at pH 3.3 and a flow rate of 1 mL/min. at 257 nm where: A: Solvent front B: Oxidative degradate (10 μg/mL) C: CIL (10 μg/mL).

Figure 6: Zero order absorption spectra overlay of 20 μg/mL CIL (a) and 20 μg/mL Oxidative degradate (b) in methanol.

Figure 7a: First derivative absorption spectra of cilostazol and oxidative degradation product in methanol showing the zero crossing point of each, where a,b,c,d. different increasing concentrations of cilostazol and e: is 20 μg/ mL oxidative degradate.

Figure 7b: First derivative absorption spectra of cilostazol and oxidative degradation product in methanol showing the zero crossing point of each, where a,b,c,d: different increasing concentrations of oxidative degradate and e: is 10μg/mL cilostazol.

Figure 8: First derivative absorption spectra of cilostazol and oxidative degradate in their synthetic mixture where: a (7 μg/mL and 3 μg/mL), b (4 μg/ mL and 6 μg/mL) and c (2 μg/mL and 8 μg/mL) of CIL and oxidative degradate, respectively.
Chromatographic conditions study
After many experimental trials, good resolved symmetrical peaks were resulted. Summary of those trials can be summarized as follows
Column selection: Symmetry® C18 column (250 mm × 4.6 mm i.d., 5 μm particle size); shim-pack VP- ODS column (250 mm × 4.6 mm i.d., 5 μm particle size), Shimadzu, Kyoto, Japan and monolithic C8 column (3 mm × 4.6 mm i.d., 2 μm pore size highly porous column) were used to evaluate chromatographic performance and achieve good separation. Experimental studies proved that monolithic column (3 mm × 0.6 mm i.d., 2 μm pore size highly porous monolithic column) was the column of choice for this study giving well defined symmetrical peaks of both studied drugs with good resolution and short retention time.
Choice of appropriate detection wavelength: UV absorption spectra of CIL/TEL against blank methanol showed that maxima for CIL was obtained at 220 nm and 260 nm and for Telmisartan at 240, 257 and 300 nm (Figure 2). It was found that 257 nm was the wave length of choice for [3] CIL and TEL showing reasonable absorbance. Therefore, The UV detection was recorded at 257 nm allowing the assay of both drugs with high sensitivity.
Choice of appropriate detection wavelength: UV absorption spectra of CIL/TEL against blank methanol showed that maxima for CIL was obtained at 220 nm and 260 nm and for Telmisartan at 240, 257 and 300 nm (Figure 2). It was found that 257 nm was the wave length of choice for [3] CIL and TEL showing reasonable absorbance. Therefore, The UV detection was recorded at 257 nm allowing the assay of both drugs with high sensitivity.
Mobile phase composition: Many modifications in the eluent constituents were done to reach the conditions which lead to high chromatographic performance. Based on peak symmetry, tailing factor and retention time, the selection process was done. These modifications included; the change of mobile phase pH, the type of the organic modifier and its ratio, phosphate buffer concentration and also the flow rate. The produced results are illustrated in Table 1.
| Parameter | No. of theoretical Plates (N) | Tailing factor (Tf) | Resolution (Rs) | Relative Retention (α) | |||
|---|---|---|---|---|---|---|---|
| CIL | TELMI | CIL | TELMI | ||||
| pH of mobile phase | 3 | 4670 | 6410 | 2.6 | 2.28 | 3.243 | 1.29 |
| 4 | 5513 | 6603 | 2.26 | 2.16 | 2.648 | 1.24 | |
| 4.5 | 5618 | 6366 | 0.085 | 0.092 | 2.671 | 1.45 | |
| 5 | 5461 | 5487 | 2.405 | 2.404 | 3.52 | 1.38 | |
| 5.5 | 5612 | 5380 | 2.3 | 2.146 | 2.557 | 1.23 | |
| ACN: buffer ratio | 70:30 | 2685 | 3264 | 0.78 | 3.59 | 1.33 | 2.62 |
| 60:40 | 4500 | 4942 | 2.4 | 2.24 | 2.619 | 1.57 | |
| 50:50 | 5618 | 6366 | 0.085 | 0.092 | 2.671 | 1.28 | |
| 40:60 | 6686 | 6444 | 2.1 | 1.97 | 5.54 | 1.55 | |
| 30:70 | 7791 | 7286 | 2.0 | 1.774 | 6.285 | 1.41 | |
| Conc. of phosphate buffer (M) | 0.01 | 7438 | 7098 | 2.056 | 1.778 | 5.66 | 1.48 |
| 0.03 | 7110 | 6580 | 1.508 | 1.738 | 5.763 | 1.52 | |
| 0.05 | 6686 | 6444 | 2.1 | 1.97 | 5.54 | 1.54 | |
| 0.07 | 6912 | 6577 | 2.129 | 2.00 | 5.37 | 1.44 | |
| 0.1 | 6746 | 6385 | 2.24 | 2.055 | 4.578 | 1.45 | |
| 0.15 | 7023 | 7551 | 2.13 | 1.768 | 5.202 | 1.42 | |
| Effect of flow rate (mL/min.) | 0.8 | 6233 | 6475 | 1.97 | 2.00 | 5.957 | 1.57 |
| 1.0 | 7110 | 6580 | 1.508 | 1.738 | 5.763 | 1.52 | |
| 1.2 | 6028 | 5790 | 1.342 | 1.554 | 5.80 | 1.35 | |
Where: Number of theoretical plates 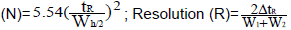 Tailing factor (Tf), Selectivity factor (α)=tr2-tm/tr1-tm |
|||||||
Table 1: Optimization of the chromatographic conditions for separation of mixture of CIL and TELMI by the proposed HPLC method.
Mobile phase pH: The mobile phase pH was studied and showed that pH 4.5 was selected as the most appropriate one providing fully symmetrical peaks with high theoretical plates count, good resolution and less tailing factor value for both drugs within a short time as illustrated in Table 1. At pH above 5.5 a great overlapping between the two peaks occurs, and the two drugs eluted at the same retention time (2.4 min) at pH 6.
Ratio of organic modifier: Acetonitrile: 0.03 M phosphate buffer (40: 60, v/v) was selected as the most suitable mobile phase, it allowed the separation of CIL and TEL within a short run time with high resolution, high sensitivity for both drugs. By decreasing the acetonitrile ratio less than 40, the peak symmetry not affected but the retention time of both drugs increase and the run time became 15 min. By elevating organic modifier ratio, overlap between the peaks of both drugs occurred.
Molar concentration of phosphate buffer: The influence of molar concentration changing of phosphate buffer on the chromatographic efficiency was studied using eluents containing concentrations of 0.01 M-0.15 M of phosphate buffer. It was found that, the molar concentration of phosphate buffer is not a critical item affecting the peak separation of both studied drugs as by changing the molar strength the peak sensitivity, resolution and asymmetry not greatly affected. 0.03M was selected as the most appropriate concentration due to highly symmetrical peaks.
Mobile phase replacement: When acetonitrile was replaced with methanol using the selected ratio methanol: phosphate buffer (40:60, v/v), no peaks were eluted till 30 min and upon replacement of phosphate buffer with water to become acetonitrile: water (40:60, v/v), the peak of TEL not appear till 30 min. So, acetonitrile: phosphate buffer (40:60, v/v) was the selected mobile phase for this study to obtain perfect chromatographic performance.
Flow rate influence: The influence of flow rate in the range of 0.8- 1.2 mL/min on the separation of both drugs peaks was studied; 1.0 mL/ min was the selected flow rate to obtain good separation in a small retention time.
Choice of internal standard: Several drugs were studied as internal standard; Dapoxitine, Clopidogrel, Dipyridamol, and Lamotrigene. Dipyridamol was selected as the most suitable internal standard with well resolved peak with no interference with peaks of both studied drugs and giving a well-defined peak with concentration (3 μg/mL).
Validation of the proposed methods
The proposed HPLC method was validated as per ICH guide lines using the following items: Linearity, specificity, accuracy, precision, LOD and LOQ.
Linearity and range: The calibration graphs for CIL and TEL assay by the developed HPLC method were produced from the peak area ratio [drug/I.S.] versus the concentration in μg/mL plots for both drugs. The high values of correlation coefficient showed high linearity for the concentration ranges stated in Table 2. Regression [3] analysis of the data yielded the corresponding equations:
P=0.084+ 0.430 C (r=0.9999) for CIL
P=0.350+0.390 C (r=0.9999) for TEL
| Parameter | CIL | TEL |
|---|---|---|
| Linearity and range (µg/mL) | (0.5-15) | (0.25-20) |
| LOD (µg/mL) | 0.04 | 0.06 |
| LOQ (µg/mL) | 0.11 | 0.20 |
| Intercept (a) | 0.084 | 0.35 |
| Slope (b) | 0.430 | 0.39 |
| Correlation coefficient(r) | 0.9999 | 0.9999 |
| S.D. of residuals (Sy/x) | 8.5 × 10-3 | 1.5 × 10-2 |
| S.D. of intercept (Sa) | 4.9 × 10-3 | 8.3 × 10-3 |
| S.D. of slope (Sb) | 6.0 × 10-4 | 8.0 × 10-4 |
| S.D. | 0.56 | 0.63 |
| %RSD | 0.55 | 0.63 |
| %Error | 0.21 | 0.24 |
Where: (LOD) Limit of detection, (LOQ) Limit of quantitation, (% RSD) Percentage relative standard deviation, (% Error) Percentage relative error.
Table 2: Analytical performance data for Cilostazol and Telmisartan assay using the proposed HPLC methods.
Where: P represents the peak area ratio, C is corresponding to the concentration of the drug in μg/mL and r refers to the correlation coefficient. Statistical analysis [41] of the results obtained from the mentioned regression equations produced high value of the correlation coefficient (r), small values of the standard deviation of residuals (Sy/x), of intercept (Sa), and of slope (Sb), and also percentage relative standard deviation and percentage error were very small value (Table 2). This analysis proved that the developed HPLC method was of high linearity.
Detection limit (LOD) and quantitation limit (LOQ): The limit of detection (LOD) was defined as the minimum level at which the drug can be reliably detected [41]. The limit of quantitation (LOQ) was determined by finding the lowest concentration that can be measured according to ICH guidlines [41,42] below which the calibration curve become nonlinear. LOD and LOQ were calculated from the following equations.
LOD=3.3 Sa /b LOQ=10 Sa /b
Where Sa=standard deviation of the intercept of the calibration curve and b=slope of the calibration curve. The acquired data of the developed HPLC method were cited in Table 2.
Accuracy and precision: By comparing the developed HPLC method assay data with those obtained by the authorized one described by the USP [3], the accuracy of the developed method was confirmed. Statistical assessment of the obtained results using Student's t-test and variance ratio F-test [42] proved that there is no significant difference between the performance of the two methods regarding the accuracy and precision, respectively (Table 3). Inter-day and intra-day assay for the studied drugs were established to indicate that the developed method was precise.
| Drug | Proposed HPLC method | Reference method [3,4] | ||||
|---|---|---|---|---|---|---|
| Amount taken (mg/mL) |
Amount found (mg/mL) |
% Found | Amount taken (mg/mL) |
Amount found (mg/mL) |
% Found | |
| CIL | 0.5 | 0.500 | 100.02 | 0.2 | 0.202 | 101.15 |
| 1.0 | 0.999 | 99.91 | 0.5 | 0.496 | 99.25 | |
| 2.0 | 2.024 | 101.24 | 1.0 | 1.001 | 100.14 | |
| 4.0 | 3.981 | 99.53 | ||||
| 8.0 | 7.972 | 99.66 | ||||
| 10.0 | 10.015 | 100.15 | ||||
| 15.0 | 15.004 | 100.03 | ||||
| Mean ± S.D. |
100.08 0.56 |
100.18 0.95 |
||||
| t-test | 0.22 (2.30) | |||||
| F-test | 2.90 (5.14) | |||||
| TEL | 0.25 | 0.248 | 99.44 | 5.0 | 5.011 | 100.24 |
| 1.0 | 0.995 | 99.55 | 8.0 | 7.970 | 99.63 | |
| 2.0 | 1.972 | 98.65 | 10.0 | 10.017 | 100.18 | |
| 5.0 | 5.005 | 100.11 | ||||
| 10.0 | 10.068 | 100.68 | ||||
| 15.0 | 14.960 | 99.74 | ||||
| 20.0 | 19.993 | 99.97 | ||||
| Mean | 99.73 0.63 |
100.02 0.34 |
||||
| ± S.D. | ||||||
| t-test | 0.72 (2.30) | |||||
| F-test | 3.52 (19.32) | |||||
N.B.*the figures between parentheses are the tabulated t and F values at P=0.05 (40)
Table 3: Assay results for the determination of CIL and TEL drugs in pure form using the proposed HPLC procedure.
Intra-day precision: Intra-day precision was performed for the developed HPLC method through replicate analysis of three concentrations of both studied drugs on three successive times within a day. Small values of % Error and % RSD were resulted from the obtained results which indicate that the developed method is of high accuracy and precision, respectively. The results are summarized in Table 4.
| Parameters | CIL concentration (mg/mL) | TEL concentration (mg/mL) | |||||
|---|---|---|---|---|---|---|---|
| 2.0 | 8.0 | 10.0 | 1.0 | 5.0 | 10.0 | ||
| Intraday | % Found | 100.85 | 99.55 | 99.40 | 99.80 | 99.16 | 99.25 |
| 99.90 | 100.03 | 100.50 | 100.55 | 100.50 | 100.40 | ||
| 99.55 | 100.44 | 99.66 | 99.20 | 99.60 | 99.68 | ||
| x ± SD |
100.10 0.67 |
100.01 0.45 |
99.85 0.58 |
99.85 0.68 |
99.75 0.68 |
99.78 0.58 |
|
| % RSD | 0.67 | 0.45 | 0.58 | 0.68 | 0.68 | 0.58 | |
| % Error | 0.39 | 0.26 | 0.33 | 0.39 | 0.40 | 0.34 | |
| Interday | % Found | 100.15 | 99.15 | 100.00 | 100.40 | 100.11 | 99.40 |
| 99.57 | 99.66 | 98.59 | 99.55 | 100.29 | 100.80 | ||
| 101.24 | 100.80 | 99.60 | 99.03 | 101.76 | 99.96 | ||
| x ± SD |
100.32 0.85 |
99.87 0.85 |
99.40 0.73 |
99.66 0.69 |
100.72 0.91 |
100.05 0.71 |
|
| % RSD | 0.85 | 0.85 | 0.73 | 0.69 | 0.90 | 0.71 | |
| % Error | 0.49 | 0.49 | 0.42 | 0.40 | 0.52 | 0.41 | |
Table 4: Precision data for the determination of CIL and TEL by the proposed HPLC procedure.
Inter-day precision: Inter-day precision was performed via replicate analysis of three concentrations of the studied drugs on three successive days. Table 4 represents the obtained results. The small values of % Error and % RSD indicate high accuracy and precision of the developed method, respectively.
Robustness of the developed HPLC method: The robustness of an analytical method is defined as the method capability to remain unaffected by deliberate minor changes in experimental parameters. The proposed HPLC method showed robustness upon minor changes of chromatographic conditions such as; mobile phase pH (4.5 ± 0.2), phosphate buffer strength (0.030 ± 0.01 M). The ratio of acetonitrile: 0.03 M phosphate buffer in the mobile phase is a critical parameter for the separation process, where small changes in organic modifier ratio from (40: 60, v/v) greatly affect both drugs resolution and run time.
Specificity: The proposed HPLC method was specific for assay of CIL and TEL in tablets. Common tablet excipients did not affect the separated peaks in the developed HPLC method.
Application of the developed HPLC method for CIL and TEL assay in their tablets
The proposed HPLC method was successfully utilized for simultaneous quantitation of CIL and TEL in their synthetic mixtures that medicinally recommended in ratio of 4:1 (w/w) (Table 5). Also, the developed method was used for the determination of the studied drugs separately each in its own tablet (Table 6). Good agreement of the resulted data with those of the authorized methods stated by USP [3] was found. Statistical analysis of the produced results using Student's t-test and variance ratio F test [42] proved that there is no significant difference between the performance of the two methods regarding the accuracy and precision, respectively.
| CIL/TEL ratio | Proposed HPLC method | Reference method | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 3 | 4 | ||||||||
| Amount taken (mg/mL) |
Amount found (mg/mL) |
% Found | %Found | ||||||
| CIL | TEL | CIL | TEL | CIL | TEL | CIL | TEL | ||
| 4:1 | 4.0 | 1.0 | 3.972 | 1.005 | 99.32 | 100.53 | 101.15 | 100.24 | |
| 8.0 | 2.0 | 8.053 | 1.989 | 100.66 | 99.46 | 99.25 | 99.63 | ||
| 12.0 | 3.0 | 11.971 | 3.005 | 99.77 | 100.18 | 100.14 | 100.18 | ||
| x ± SD |
99.92 0.68 |
100.06 0.55 |
100.18 0.95 |
100.02 0.34 |
|||||
| %RSD | 0.68 | 0.54 | 0.94 | 0.33 | |||||
| %Error | 0.39 | 0.31 | 0.54 | 0.19 | |||||
| T | 0.38 | 0.11 | |||||||
| F | 1.94 | 2.63 | |||||||
N. B. Each result is the average of three separate determinations.
The values of tabulated t and F tests are 2.78 and 19.00, respectively at p=0.05 [29].
Table 5: Assay results for the determination of the CIL and TEL in synthetic mixtures in ratios of 4:1 (w/w) by the proposed HPLC method.
| Tablet | Proposed method | Official method 3 | ||||
|---|---|---|---|---|---|---|
| Amount taken (mg/mL) |
%Found | Amount taken (mg/mL) |
%Found | |||
| Claudol Tablet (100.00 mg CIL) |
1.0 | 98.86 | 0.2 | 101.65 | ||
| 4.0 | 100.42 | 0.5 | 98.94 | |||
| 10.0 | 99.94 | 1.0 | 100.20 | |||
| Mean | 99.74 0.80 | 100.26 1.36 | ||||
| ± S.D. | ||||||
| t-test | 0.57 (2.77) | |||||
| F-test | 2.88 (19.0) | |||||
| Official method 3 | ||||||
| Amount taken (mg/mL) |
%Found | Amount taken (mg/mL) |
%Found | |||
| Micardis Tablet (40.00 mg TEL) |
1.0 | 100.96 | 5.0 | 100.27 | ||
| 5.0 | 99.64 | 8.0 | 99.58 | |||
| 10.0 | 100.07 | 10.0 | 100.20 | |||
| Mean | 100.22 0.67 | 100.02 0.38 | ||||
| ± S.D. | ||||||
| t-test | 0.46 (2.77) | |||||
| F-test | 3.14 (19.0) | |||||
N.B.*each result is the average of three separate determinations.
*The figures between parentheses are the tabulated t and F values at P=0.05 [40].
Table 6: Assay results for the determination of both drugs in their tablets, separately.
Application of the developed HPLC method for assay of cilostazol with existence of its oxidative degradate
The proposed HPLC method was applied for CIL assay and its oxidative degradate, producing successful results. The results of assay were abridged in Table 7. Under the optimized experimental conditions, linearity of the method was achieved by plotting the peak area against the concentration in μg/mL for CIL and oxidative degradate, simultaneously. Table 8 showed assay results of 10 μg/mL CIL different times in presence of different percentages of oxidative degradation product. The regression equations were as follows:
P=9.09+38.53 C (r=0.9999) (Cilostazol)
P=-7.72+21.32 C (r=0.9999) (Oxidative degradate)
| Item | Synthetic mixture of cilostazol and its oxidative degradation product | |||
| Amount taken (mg/mL) |
%Recovery | |||
| CIL | Oxidative degradate | CIL | Oxidative degradate | |
| 8 | 2 | 99.85 | 99.03 | |
| 7 | 3 | 100.06 | 100.58 | |
| 6 | 4 | 100.13 | 100.30 | |
| 4 | 6 | 100.23 | 99.86 | |
| 2 | 8 | 99.53 | 99.99 | |
| x | 99.96 | 99.95 | ||
| %RSD | 0.28 | 0.59 | ||
Table 7: Assay results for determination of Synthetic mixture of Cilostazol and its oxidative degradation product using the proposed HPLC method.
| CIL | Oxidative degradate | %Recovery | |
|---|---|---|---|
| CIL | Oxidative degradate | ||
| (10 mg/mL) | 10% | 100.03 | 101.70 |
| 20% | 98.13 | 100.05 | |
| 40% | 98.40 | 99.65 | |
| 60% | 99.45 | 99.40 | |
| 80% | 99.09 | 100.40 | |
| x | 99.02 | 100.24 | |
Table 8: Assay results for determination of oxidative degradation product in presence of 10 μg/mL CIL using the proposed HPLC method.
Where: P represents the peak area, C is the concentration in μg/mL and r refers to the correlation coefficient. The results obtained indicated the high linearity of the calibration curve performed for determination of cilostazol and oxidative degradation product.
First derivative spectrophotometric method for assay of cilostazol and its oxidative degradation product
The calibration plots for the determination of CIL and its oxidative degradate by the proposed derivative spectrophotometric method were obtained by plotting the amplitude of the derivative peaks against the concentration in μg/mL. Table 9 shows the linearity range of CIL and its oxidative degradate using derivative spectrophotometricmethod. The following equations illustrate the regression analysis of data:
1D228=-5 × 10-4+6.5 × 10-3C (r=0.9999) for (Cilostazol)
1D257=0.05+8.7 × 10-3C (r=0.9999) for (Oxidative degradate)
| Parameter | CIL | Oxidative degradate |
|---|---|---|
| Linearity and range (µg/mL) | (1.0-35) | (2.0-50) |
| LOD (µg/mL) | 0.23 | 0.41 |
| LOQ (µg/mL) | 0.71 | 1.23 |
| Intercept (a) | -5 × 10-4 | 0.05 |
| Slope (b) | 6.5 × 10-3 | 8.7 × 10-3 |
| Correlation coefficient(r) | 0.9999 | 0.9999 |
| S.D. of residuals (Sy/x) | 7 × 10-4 | 1.4 × 10-3 |
| S.D. of intercept (Sa) | 5 × 10-4 | 1.1 × 10-3 |
| S.D. of slope (Sb) | 0.00 | 0.00 |
| S.D. | 0.60 | 0.37 |
| %RSD | 0.60 | 0.37 |
| %Error | 0.25 | 0.15 |
Table 9: Analytical performance data for Cilostazol assay in presence of its oxidative degradation product using derivative spectrophotometric method.
Where: (D wavelength) is the amplitude of the first derivative spectra at the stated wave length, C is the concentration in μg/mL and r is the correlation coefficient. Also, Table 10 shows assay results of CIL and its oxidative degradate in their synthetic mixture using derivative spectrophotometric method.
| Item | Proposed HPLC method | |||||
|---|---|---|---|---|---|---|
| Amount taken (mg/mL) |
Amount found (mg/mL) |
%Found | ||||
| CIL | Degradate | CIL | Degradate | CIL | Degradate | |
| 7.0 | 3.0 | 6.986 | 2.987 | 99.81 | 99.59 | |
| 4.0 | 6.0 | 4.013 | 6.036 | 100.34 | 100.61 | |
| 2.0 | 8.0 | 1.986 | 7.987 | 99.33 | 99.85 | |
| x ± SD |
99.83 0.51 |
100.02 0.53 |
||||
| %RSD | 0.50 | 0.53 | ||||
| %Error | 0.29 | 0.31 | ||||
Table 10: Assay results for the determination of the CIL and its oxidative degradate synthetic mixtures using derivative spectrophotometric method.
Very simple, rapid, accurate and precise HPLC method was developed for simultaneous determination of CIL and TEL. The method was applied to the analysis of both drugs in their pure and tablet formulation without interference from common excipients. The results obtained were of good agreement with the authorized method. The developed HPLC method was accurate and highly precise with low percentage error and relative standard deviation so, it could be applied in quality control laboratories. HPLC method was applied for a selective assay of cilostazol in presence of its oxidative degradate with good linearity for both. Also selective first derivative spectrophotometric method was applied for assay of cilostazol in presence of its oxidative degradate showing high linearity.