Fungal Genomics & Biology
Open Access
ISSN: 2165-8056
ISSN: 2165-8056
Research Article - (2014) Volume 4, Issue 1
Keywords: Magnaporthe oryzae, AvrPiz-t, Transposon insertion, SNP, Gene diversity
Most plants are not affected by the majority of microbes they encounter because of their functional defense system. During the coevolution of plants and phytopathogens, plants have evolved multiple branches of defenses including one specific reactions triggered by pathogen delivered effectors [1,2]. According to the “gene-for-gene” model proposed by Flor in 1971, plants developed a mechanism to detect avirulence (Avr) gene products via corresponding resistance genes (R genes), which results in a subsequent hypersensitive reaction (HR) to suppress microbial growth [3].
Magnaporthe oryzae is an ascomycete fungus and the causal agent of rice blast disease, which is considered as one of the most devastating diseases of rice with regard to grain loss. This disease has been found in over 85 countries and the amount of rice lost each year could feed upward of 60 million people [4], as severe disease breakouts may lead to as much as 90% yield loss in a field or region [5].
In order to provide a genetic based disease control and understand molecular interactions between plants and fungal pathogens, great effort has been made to identify and clone R genes of rice cultivars and Avr genes in M. oryzae. To date, a large number of R genes have been successfully transferred into elite rice cultivars to create resistance rice lines through breeding programs [6]. However, R gene mediated resistance can be rapidly broken down after several generations [5], which is hypothesized to be result of the inherent instability and high variation of M. oryzae Avr genes especially those in subtelomeric regions [7]. For example, while Piz-t serves as a proven R gene in rice blast disease control, its effectiveness is challenged by the fast evolution of the AvrPiz-t locus, which was cloned in 2009 [8]. In one survey we performed in major rice growing areas in China, although the Piz-t gene remains functional in majority areas (Figure 1), the virulence frequency of AvrPiz-t containing strains from two disease nurseries in Fujian province has increased from 12.5% to 100% within 14 years (Figure 2). So far, several Avr genes have been cloned in M. oryzae, including AVR-Pita [9], AVR-CO39 [10], PWL1 [11], PWL2 [12], ACE1 [13], AVR-Pia, AVR-Pii, and AVR-Pik/km/kp [14]. Interestingly, it was found that some AVR genes of M. oryzae contain higher level of variations compared to other regions of genome. As exampled by the AVR-Pita1 gene, its ORF showed high polymorphisms among 30 Thai rice blast isolates resulting in varying pathotypes. It was also found that the AVR-Pita1 gene locus in the Thai blast isolates were under a positive selection pressure [15]. Similarly, the AVR-Pita1 alleles in field isolates of the USA were found to be under positive selection. Consequently, mutations at the coding regions resulting amino acid alteration, deletion resulting in frame shift and transposon insertion at a critical motif of the gene have been found to be responsible for defeating the corresponding Pi-ta R gene [16-18]. These findings suggest that M. oryzae has developed sophisticated mechanisms to survive and retain virulence and that these mechanisms may be different between genes.
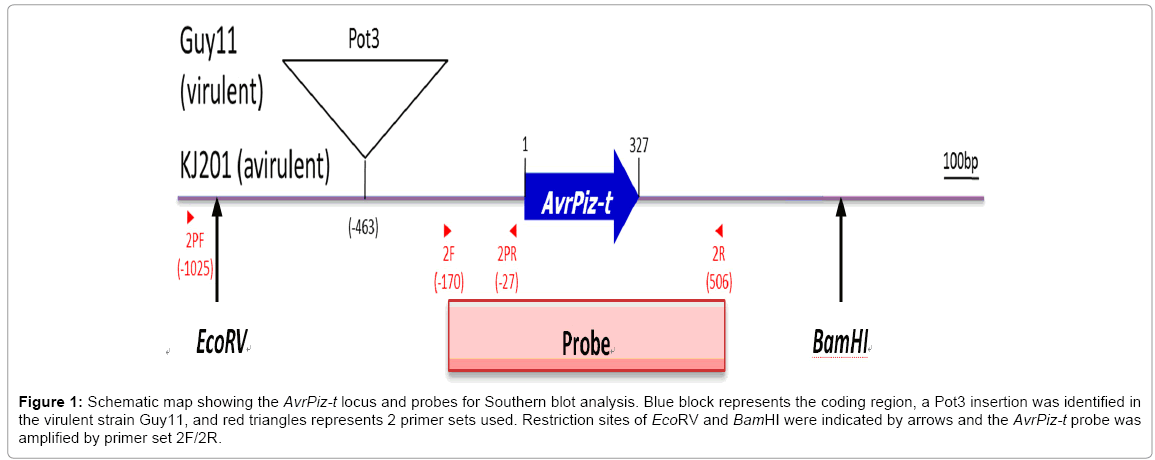
Figure 1: Schematic map showing the AvrPiz-t locus and probes for Southern blot analysis. Blue block represents the coding region, a Pot3 insertion was identified in the virulent strain Guy11, and red triangles represents 2 primer sets used. Restriction sites of EcoRV and BamHI were indicated by arrows and the AvrPiz-t probe was amplified by primer set 2F/2R.
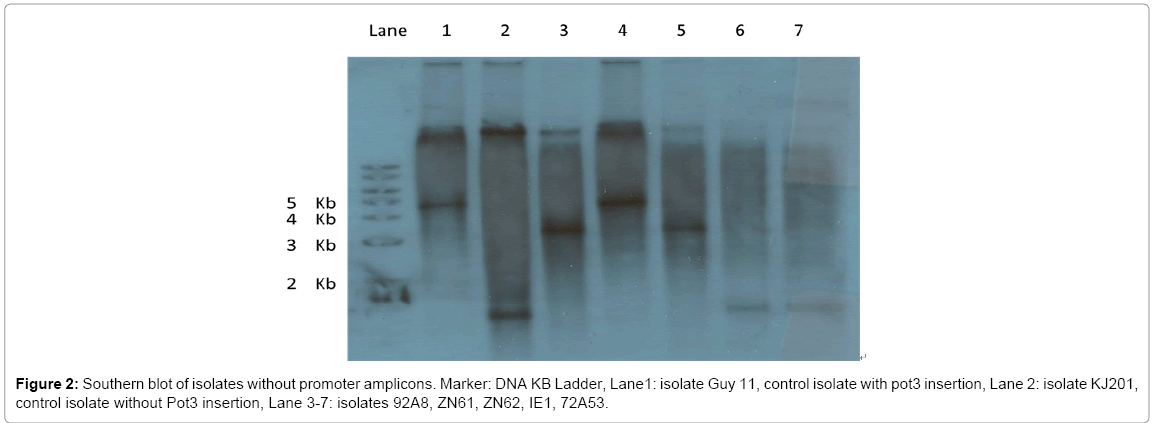
Figure 2: Southern blot of isolates without promoter amplicons. Marker: DNA KB Ladder, Lane1: isolate Guy 11, control isolate with pot3 insertion, Lane 2: isolate KJ201, control isolate without Pot3 insertion, Lane 3-7: isolates 92A8, ZN61, ZN62, IE1, 72A53.
AvrPiz-t was cloned in 2009 using a map-based strategy and encodes a 108-amino-acid predicted secreted protein [19]. The structure of the AvrPiz-t protein has recently identified based on Nuclear Magnetic Resonance (NMR) measurements, which forms a six-strand β-sandwich structure, while both N- and C-termini are disordered [20]. The mechanism of its function has also been explored in a recent study: during the infection process, AvrPiz-t accumulates in a specific structure called biotrophic interfacial complex (BIC) and is then transported into rice cells [21]. In the susceptible rice host, AvrPiz-t suppresses the reactive oxygen species (ROS) generation and thus strengthens pathogenicity in cultivars without Piz-t R gene [22].
It was proposed by Jones and Dangl [2] that AVR genes are usually flanked by transposable elements (TEs) or telomeres, which may serve as a simple strategy for pathogen to evade host detection. In M. oryzae, it has been reported that AVR genes are sometimes tightly associated with diverse TEs [13,23,24] in either the promoter or coding regions that can alter strain virulence, as been reported in Avr-Pita [16,25]. For instance, functional Avr-Pita homologs were found located in different chromosomes in a M. oryzae population survey, and were accompanied by a variety of TEs such as Inago1, Inago2, Pyret, Pot2, and Pot3. Based on the strong association between the Avr-Pita translocation events and the flanking TEs, the authors suggested that the inserted TEs may be essential for the mobility of Avr-Pita [26]. In M. oryzae strain GUY11, a virulence strain to Piz-t containing cultivar, AvrPiz-t was found present but having a transposable element Pot3 inserted in its promoter region [8]. In the 146 kb AvrPiz-t locus of the strain 70-15 which is derived from GUY11, TEs sequences were also found for as much as 43.2% of the genomic content at this locus, suggesting the dynamic evolution in this region [27].C
To evaluate the diversity of the AvrPiz-t gene, and determine its effect to virulence, we characterized 711 M. oryzae field strains in total: 313 isolates from different geographic origins (38 countries), which are referred to as the “Global strains” group, and an additional 398 isolates from different regions in China, which are referred to as the “Chinese strains” group. Most of the strains were isolated from rice but that some “non rice” strains were also included. In this study, we PCR amplified AvrPiz-t ORFs as well as promoter regions of each strain, and then compared the sequence and structure variation using Sanger sequencing and Southern hybridization. Strains with polymorphisms in the ORF were classified into groups based on mutation type and site. Pathogenicity assays were applied to evaluate the association between AvrPiz-t gene/promoter polymorphism and virulence.
Fungal isolates
Fungal isolates used in this study were collected through collaborating labs from over 38 countries and regions worldwide: China, Egypt, India, The Philippines, Burundi, Brazil, Ivory Coast, Colombia, Cameroon, South Korea, France, Gabon, French, Guyana, Hungary, Japan, Kenya, Morocco, Madagascar, Mali, Portugal, Russia, Rwanda, Spain, Thailand, USA and Vietnam. Figure 4 shows the world map where these isolates were collected. Another batch of field isolates collected from different regions of China was added into analysis. Single spores were isolated from each of these strains and plated on complete media agar plates (0.75% yeast extract, 0.75% casamino acid, 0.1% sucrose and 1% agar) covered with desiccated filter paper, which were then incubated at room temperature for 7 days. Fungal mycelia were stored on filter papers at -20°C.
DNA preparation
Genomic DNA was extracted using an in-house method optimized for fungal DNA. Isolates were transferred and grown in liquid complete media at room temperature in dark condition with shaking for 7 days. Mycelia were collected using funnel, squeezed to get rid of water and frozen dried overnight. Dried mycelia were grounded into powder with a pestle and petrol. One quarter of 2 ml tube was filled with mycelia powder and mixed with DNA extraction buffer (100 mM Tris-HCl pH=8.0, 100 mM EDTA pH=8.0 and 250 mM NaCl) and proteinase K, followed by incubating at 50 oC for 1 hr. 100 ul of 10% N-laurylsarcosyl was then added and incubated at 55°C for 1hr. Supernatant containing genomic DNA was obtained by centrifuge for 15 mins at 5,000 rpm at room temperature. Genomic DNA was extracted and purified using phenol-chloroform extraction method [28]. Final concentration of genomic DNA was adjusted to 50 ng/uL.
PCR amplification and sequencing
The ORFs and promoter regions of AvrPiz-t were amplified from genomic DNA of each Global strains with 2F/2R and 2PF/2PR primer pairs respectively, while for Chinese strains 9F/10R and 13F/14R were used (Table 1). Taq PCR kit (New England Biolabs, Inc., MA, USA) was used to perform 20 μl PCR reactions: 2 ul genomic DNA (25 ng), 2 ul thermo-buffer, 1 ul dNTP (400 mM of each dNTP), 1 ul of 10 uM forward primer, 1 ul of 10 uM reverse primer, 0.5 ul Taq and 12.5 ul distilled H2O. Reactions were performed using the following cycle: 95°C for 3min, 25 cycles of 95°C for 1 min, 55-60°C for 30 seconds (varies with different primer pairs) and 72°C for 1 min, followed by a final extension step at 72°C for 7 min. The size of amplified fragment was estimated by electrophoresis gel with 1kb DNA ladder. All amplicons were purified using QIAquick PCR Purification Kit (Qiagen Inc., CA, USA) and sent for Sanger sequencing with 2F/2R and 2PF/2PR for coding region and promoter region respectively from both forward and reverse direction.
| Haplotypes | Positions | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 27 | 56 | 68 | 80 | 120 | 156-159 | 178 | 192 | 238 | 325 | |
| I | T/C | |||||||||
| II | G/T | |||||||||
| III | G/C | |||||||||
| IV | G/A | |||||||||
| V | G/T | G/A | G/C | AGGG/CAAC | G/C | |||||
| VI | G/A | G/C | AGGG/CAAC | G/C | ||||||
| VII | A ins | |||||||||
| VIII | T del | |||||||||
Table 1: The SNP, insertion, and deletion identified in AvrPiz-t coding region and their positions. Isolates were categorized into 8 different haplotypes based on the comparison to sequence from strain KJ201.
Southern hybridization
Genomic DNA of selected isolates was digested with EcoRV and BamHI restriction enzymes (New England Biolabs Inc., USA). The digestion scheme diagram was shown in Figure 2. Digested genomic DNA was purified using phenol-chloroform method and electrophoresed on a 1.5% agarose gel at 40 voltages overnight to separate fragments by size. Fragments were transferred onto Hybond N+ membrane by capillary blotting. Probe was amplified with primer pair 2F and 2R, labeled and hybridized onto DNA membrane. Signals were detected on CXS high speed blue film. All performances were followed by the instruction of Amersham ECL Direct Labeling and Detection System kit (GE Healthcare Life Scienses, PA, USA).
Rice cultivar and pathogenicity assay
To evaluate virulence of AvrPiz-t polymorphic isolates on different hosts, 7-8 week-old rice cultivar Toride (for Global strains) or IRBL11 (for Chinese strains) containing Piz-t, and Nipponbare lacking Piz-t were used in this study. Conidia of selected strains were harvested from 7-day-old V8 juice agar plates and suspended in 250 ppm Tween 20 with a concentration of 5×105 conidia per ml. Ten ul conidial suspension of each strains were dropped onto punctuated rice leaves and wrapped with transparent tape, followed by incubation at 25°C in dark and moisture incubator. Disease severity was evaluated 7 days after inoculation using the rating system descript by Valent [29].
Data mining and sequence analysis
ORFs of the AvrPiz-t gene in each isolates were assembled and aligned by CLC sequence viewer 6.0 (CLC bio Inc.). Haplotypes, polymorphic sites, sliding window analysis and natural selection test were performed by DnaSP 5.0 [30].
Four primer sets were used in this study (Table 1). Two primer sets were used for global strains ORF amplification and promoter amplification (Figure 1), and another two primer sets were designed and used specifically for Chinese strains. Out of the total 711 isolates, 606 had their AvrPiz-t promoters successfully amplified and 637 had their full ORFs amplified. The possible reason for the failure of PCR amplification may be due to either the high rate of polymorphisms occurring in the primer regions, or deletions/insertions, which greatly alter the length of amplified region. The sizes of promoter amplicons were compared between isolates via agarose gel electrophoresis to identify the existence of transposons. To analyze for polymorphisms in the coding region, selected ORF amplicons were subjected to Sanger sequencing
Sequence diversity in the promoter region
The promoter regions of Chinese strains showed a higher PCR amplification rate (394 successes out of 398) compared to the amplification rate of Global strains (212 successes out of 313). Based on the analysis of the amplified promoter regions in, 287 isolates out of 394 Chinese strains and 197 out of 212 Global strains showed the same size as reference avirulent strain KJ201. In the 15 Global strains with different size, 4 isolates including GUY11 contain a 4 kb promoter region, and 11 isolates contain a 5.8 kb promoter region.
As shown in the work of Li et al. [19], a Pot3 transposon inserted in the promoter region of this gene was detected for GUY11. Sequencing of random picked samples from Chinese strains validated at least two types of insertions: a “retrotransposon Inago2” insertion at 41 bp upstream ORF, and a 1870 bp insertion at 462 bp upstream the ORF.
Southern hybridization validation of transposon insertions
There were 101 Global strains that failed in amplification of their promoter regions. The failure may be due to transposon insertion in the promoter region that interrupts in the primer binding sequences, or to large transposon element insertion that makes the region length exceed PCR limit. In order to verify the transposon insertion in the promoter regions for these isolates, Southern hybridization was designed using a probe covering the AvrPiz-t coding region and 170 bp upstream promoter sequences (Figure 1). This analysis was applied to Global strains that have ORF amplicon but failed in promoter amplification. Three strains, 92A8, ZN61, ZN62, were randomly picked for validation, while KJ201, GUY11 and two other strains IE1 and 72A53 were chosen as controls. As shown in Figure 2, there is a 3 kb size difference between GUY11 and KJ201 controls in their promoter region. The two control strains IE1 and 72A53 which were known to have no TE insertion showed the same product size as KJ201. ZN61 showed the same size hybridizing band as Guy11, while 92A8 and ZN62 showed a 2 kb insertion compared to KJ201.
Polymorphisms in ORF region
The low diversity in the AvrPiz-t ORF region has been confirmed in this study. In Chinese strains, more than half of the isolates collected from Taiwan showed a 1858 bp insertion in coding region at 211 bp downstream of the start codon (Figure 3). This long insertion altered the structure of the AvrPiz-t protein as well as its function, which has been demonstrated to gain virulence to Piz-t containing rice in pathogenicity assay.
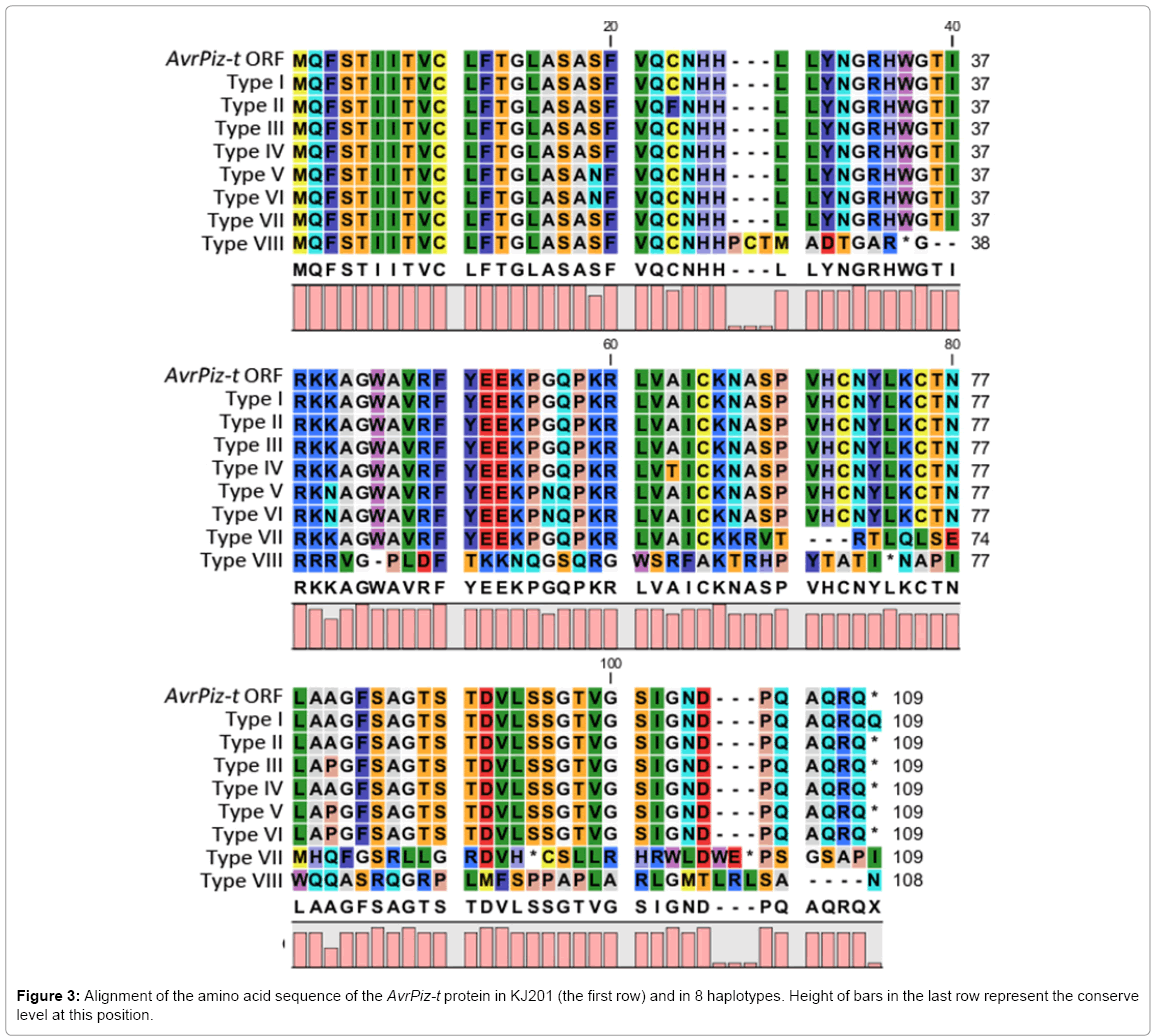
Figure 3: Alignment of the amino acid sequence of the AvrPiz-t protein in KJ201 (the first row) and in 8 haplotypes. Height of bars in the last row represent the conserve level at this position.
Of all the 243 Global strains with ORF sequenced, only 14 strains showed polymorphisms in the ORF region compared to the KJ201 gene sequence. These 14 AvrPiz-t sequences represented 8 different haplotypes. Table 1 and Figure 3 showed the 8 haplotypes of SNPs and protein differences, respectively. All DNA polymorphisms led to altered protein sequences except the SNP at 27 bp and 120 bp. Frame shift occurs in haplotypes VII and VIII, thus the sequences of the entire protein changed after the inserted/deleted position and the proteins are expected to lose function. Table 2 shows hosts and country of origin the 14 strains. Haplotypes V and VI are very similar to each other except that there is an additional polymorphism site 27 in haplotype V. Another common polymorphism site among haplotypes III, V and VI is at position 238 with a G to C substitution and the absence of nucleotide insertions or deletions. In the survey, three fungal strains belong to haplotype III, five fungal strains belong to type VI and one strain belongs to type V, which were collected from Asia, Africa and South America, respectively. Each has hosts that belong to weed species except one mainly infects Zea mays in haplotype VI. The remaining four isolates collected from Africa, Asia, North and South America on rice are classified into four different polymorphism types. Isolates with a grass host were separated from isolates that can infect rice based on the polymorphism similarity in AvrPiz-t coding region.
| Haplotypes | Strains | Countries | Host |
|---|---|---|---|
| I | BD0024 | Burundi | Oryza sativa |
| II | FC23 PH0078 |
Columbia Philippines |
Oryza sativa Oryza sativa |
| III | PH0059 PH0075 VT0032 |
Philippines Philippines Vietnam |
Leersia hexandra Brachiaria nutica Leersia hexandra |
| IV | PH0118 | Philippines | Oryza sativa |
| V | KN0006 | Kenya | Hordeum vulgare |
| VI | BR0032 BR0062 CD0155 GN0001 IN0113 JP0028 JP0038 JP0048 KN0001 |
Brazil Brazil Ivory Coast Gabon India Japan Japan Japan Kenya |
Triticum Eleusine indica Eleusine indica Zea mays Eleusine sp. Eragrostis curvala Eragrostis curvala Hordeum vulgare Hordeum vulgare |
| VII | 49D | US | Oryza sativa |
| VIII | EG85 | Egypt | Oryza sativa |
Table 2: Isolates in different haplotypes with their hosts and collected countries.
Notably, three interesting findings can be summarized from the table: 1) Three out of eight haplotypes are detected in non rice strains only; 2) Rice strains do not share haplotypes with non-rice strains; 3) Haplotype VI is shared by distant strains in the phylogeny.
Natural selection force assay
The nucleotide diversity level was calculated based on the ORF sequences from Global strains. To estimate the number of nucleotide diversity per site [31,32], π (π=0.0023) and θ (θ=0.014) were measured on the entire AvrPiz-t ORF. The π value is lower than that of the reported AVR-Pita1 sites within Thai isolates population (π=0.00891) [17]. A sliding window analysis of π value across the entire coding region was performed, as shown in Figure 4. A major peak can be found from 150-160bp in the variation distribution of AvrPiz-t alleles.
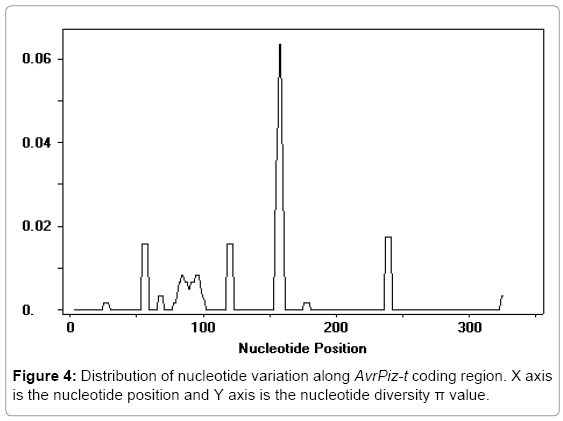
Figure 4: Distribution of nucleotide variation along AvrPiz-t coding region. X axis is the nucleotide position and Y axis is the nucleotide diversity π value.
To investigate whether the AvrPiz-t gene is under directional selection force, its ORF sequence was examined with three statistical parameters: Tajima’s D [33], Fu and Li’ D and Fu and Li’s F [34]. As shown in Table 3, the value of these three indicators is -2.307 (p<0.01), -6.010 (p<0.01) and -5.449 (p<0.01). The negative value indicates that this locus is undergoing a directional natural selection. To further determine the direction of selection power, the ratio of nonsynonymous and synonymous rate was calculated, with the assumption that ratio equals 1 indicating neutral selection. The ratio calculated for AvrPiz-t πMnon/πsyn = 1.286, which is greater than 1, indicates that AvrPiz-t is under a positive selection.
| Coding sites | Ha | Sb | π | θ | Tajima’s D | Fu and Li’s D | Fu and Li’s T |
|---|---|---|---|---|---|---|---|
| Coding | 8 | 28 | 0.0023 | 0.014 | -2.307* | -6.101* | -5.449* |
a: Number of haplotypes
b: Number of polymorphic (segregating) site, number of polymorphic (segregating) si’s estimator,
*: significant statistics < 0.01
Table 3: Polymorphism test of AvrPiz-t coding region among population.
Pathogenicity assays
Identified polymorphisms in either the AvrPiz-t promoter or coding regions may alter its function and thus change pathogenicity. To evaluate the effect of insertion on protein function, all the Chinese strains were used for pathogenicity assays (see inoculation sheet for detailed results). It can be summarized from the results that while all the avirulent strains contain wild type AvrPiz-t promoter and coding regions, 72.8% (107 of 147) of the virulent strains contain size variation in promoter regions, 21.8% (32 of 147) contain insertions in coding regions, 2.7% (4 of 147) failed in promoter or coding region amplifications, and 2.7% (4 of 147) showed no obvious size variations. The fact that majority of virulent mutant strains comes from the promoter size variation group suggests that TE insertion in promoter region plays an important role in the ability for M. oryzae to break Piz-t gene resistance in host.
In addition to insertions, SNPs observed in coding regions may also affect protein function. To estimate the effect of AvrPiz-t ORF mutation to the fungal virulence, several strains from different haplotypes were chosen for pathogenicity assays, including strain BD0024 (Type I), FC23 (Type II), 49D (Type VII), and EG85 (Type VIII). The four strains, as well as negative control strain KJ201, were inoculated on rice cultivar Nipponbare (NPB) and the Piz-t harboring cultivar Toride. Figure 5 shows that NPB is sensitive to KJ201, which induces a hypersensitive reaction on rice cultivar Toride. EG85 and FC23 can overcome resistance mediated by Piz-t and established growth on Toride, while 49D cannot infect either of these two cultivars. According to the DNA sequences at the AvrPiz-t locus of these strains, a ‘T’ insertion caused frame shift in the EG85 ORF and an early stop codon resulting in a truncated 36 amino acid protein. This suggests that this truncated protein is not a functional factor and cannot trigger host defense. A single base mutation in the FC23 ORF led to an amino acid change close to the N terminal suggesting that this region may contain a core recognition signal or is critical for maintaining the protein structure. In addition, an ‘A’ insertion also occurred in the 49D ORF but closer to the C terminal ending up with a truncated 88 amino acid protein suggesting that the functional recognition signal is close to the N terminus.
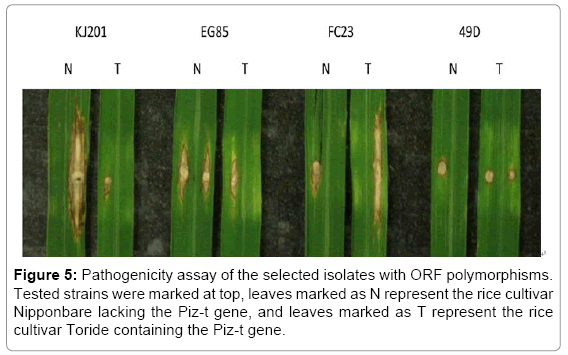
Figure 5: Pathogenicity assay of the selected isolates with ORF polymorphisms. Tested strains were marked at top, leaves marked as N represent the rice cultivar Nipponbare lacking the Piz-t gene, and leaves marked as T represent the rice cultivar Toride containing the Piz-t gene.
Strain BD0024 was also tested for pathogenicity and showed avirulence result (Data not show). These results suggest that some modifications in the protein may not alter avirulence (ie recognition) or that other avirulence genes in this strain triggered Toride 1 resistance (which may have several R genes).
Successful plant defense relies on recognition and interaction with pathogen effectors by R genes. The detailed molecular mechanisms describing how pathogens overcome specific resistance gene defense is not well understood, but it is certain that the instability of AVR genes leads to the gain of virulence. To better understand this association, genome organizations at AVR gene loci have been investigated. For instance, the AvrPi-ta1 gene in M. oryzae has been well studied for years. It has been demonstrated to interact with its cognate Pi-ta gene in rice directly [35,36], and one study shows that coding region polymorphisms exist in 5 out of 11 M. oryzae field isolates collected in China [37]. In another study on AVR-Pita1 alleles of 151 US isolates, 26 haplotypes were identified in the coding region based on DNA sequencing [17]. The high genetic diversity of the AVR-Pita1 locus is well supported by the fact that it is located in the subtelomeric region, a highly unstable region [26,38]. In addition, several other AVR genes have been mapped near the telomeric region including Avr-Pii, AVR-Pia, Avr-Pit, Avr1-Ku86, Avr1-MedNoi and PWL1 [11,39-41]. Another example on the promoter of Avr-CO39 showed that repetitive sequences including REP1, RETRO5 and MGR691/MGR508 were identified at the 5’ terminus of the Avr-CO39 locus in M. oryzae [24]. In this study, we conducted similar analysis, and 8 ORF haplotypes were identified among 243 sequenced isolates, in which haplotypes II and VIII showed gain of virulence in the pathogenicity assays compared to avirulent isolate KJ201, suggesting the possible association of virulence with DNA sequence diversity in the coding region. The coding region diversity is lower compared to the previous report of AVR-Pita1 locus. However, the result is consistent with the study conducted by Yoshida et al., who found that the majority (78%) of 1032 analyzed the AVR loci are identical in field isolates [42].
The AvrPiz-t gene resides in a region that comprises high transposable element density on chromosome 7 [43]. It was further identified that a Pot3 transposable element inserted in the 500 bp upstream region in the virulent isolate GUY11 compared to the avirulent isolate KJ201 [27]. In this study, the fact that more diversity has been identified in the promoter region of the AvrPiz-t locus compared to its coding region suggests the function of this gene in a population may be affected from the present/absent polymorphism of transposable element more often than from variation in its coding sequence. In addition to transposon elements inserted in AvrPiz-t promoter region in this study, a Pot3 element was shown to be present in the promoter region and coding region of the AVR-Pita1 gene in two different virulent isolates [25]. Also, a 1.9 kb MINE element was identified in the exon region of AVR gene ACE1 in a virulence isolates [13]. Combined with the similar findings in other fungi isolates and the AVR loci, transposon elements insertion close or inside coding region of AVR loci are crucial mechanisms the fungi use to protect themselves from being recognized by R genes.
Statistical analysis of the selection force on AvrPiz-t suggests that this gene is under positive selection and has favored amino acid substitution in its coding region. This finding is consistent with other studies that AVR genes are prone to change in positive selection [17,42,43]. Given the dynamic nature of telomeric regions, repetitive elements and transposon elements, the fact that many AVR genes are associated with these regions may provide fungi advantages to modify AVR genes adapting to R genes. For example, multiple AVR haplotypes of AVR-Pita1 were found in natural populations where only one resistant haplotype of the cognate R gene Pi-ta was found suggesting that AVR-Pita1 engages trench warfare with Pi-ta.
Now we confirmed the diversity of AvrPiz-t and the connection between sequence variance and pathogenicity, a potential method to apply this finding is design biomarkers for AvrPiz-t gene that can be used to determine the virulence of the tested strain to corresponding R gene. This can serve as an approach to estimate filed population containing functional AVR or nonfunctional alleles and deploy Piztbased resistance effectively.
In conclusion, we suggest that the AvrPiz-t alleles in world field pathogen populations have been undergoing a relatively strong positive selection that prefers amino acid substitution. The coding sequence is dynamic and transposon element insertion in the promoter region or the coding region enable M. oryzae to prevent them from being recognized by cognate R genes in their hosts.
We thank all investigators who collected fungal isolates and supported this study around the world, and Mr. Jian Chen and Ms. Qinying Qu for analyzing samples. This work was supported the Ohio Agricultural Research and Development Center and by 973 (Project No. 2012CB114001), NSFC (No. 91231121 and No. U1305211). This work was also supported by the IRMA grant (ANR-07-GPLA-007) from the Génoplante program and the GEMO project (ANR-09-GENM-029) from the ANR-Génomique program. The U.S. Department of Agriculture is an equal opportunity provider and employer.