Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2016) Volume 9, Issue 11
The existence of polypeptide chain fragments in which identical sequences translate into different secondary folds gives rise to questions concerning the structural variability associated with amyloidogenesis. In this paper the structural contribution of identical sequences to a common hydrophobic core is assessed on the basis of the fuzzy oil drop model. The model compares the observed hydrophobicity density distribution in a protein molecule to its idealized counterpart, where all hydrophilic residues are exposed on the surface while all hydrophobic residues are internalized. The conformational variability of such fragments is thought to be associated with their role: they either participate in the formation of a stable core, or become involved in mediating the protein’s biological function. The fuzzy oil drop model provides clues as to the role of chameleon sequences in prions, seen as potential loci of conformational changes resulting in amyloidogenesis.
Keywords: Chameleon sequences; Amyloidosis; Prions; Hydrophobic core; Stabilization β-sheet
The main question of structural biology: How the proteins fold reaching biologically active structural forms? Got recently extended to: How the proteins adopt the structural forms improper for biological function in amyloids? The role and status of so called “distorted” helices (polypeptide chain fragments adopting helical forms despite their high β-structure propensity) in context of chameleon sequences in relation to amyloidosis is the object of analysis. The participation of selected fragments in the structure of hydrophobic core is the criterion to characterise the specificity of these fragments. The analysis of hydrophobic core (using the fuzzy oil drop model assuming the structure of hydrophobic core as following the 3D Gauss function-maximum value of the function is located in the center of the ellipsoid with decrease as the distance versus the cented increases reaching zero level on the surface) reveals that chameleon sequences participating in distorted helices appear to fit well to the idealized hydrophobicity distribution. The question: Why do they transform to the amyloids? Remains still open. At least the fuzzy oil drop model so far does not reveal the source of the amyloidogenesis since no reason for these fragments to transform their helical structure to β-forms was found.
Protein folding, i.e., the process by which proteins attain a suitable structure, facilitating their function, is invariably tied to the following question: why do some proteins misfold? The notion that the amino acid sequence of a protein determines its 3D conformation is thus given a new meaning. Improper folding results in pathological proteins and leads to a variety of medical conditions jointly referred to as misfolding diseases [1-14]. Among such proteins are prions-misfolded structures characterized by major conformational changes which produce multimeric assemblies of β-sheets, resulting in highly deleterious health effects [15-20].
The scope of pathological effects generally depends on the location of amyloids and the specific tissue or organ which is affected by their presence. The most severe symptoms are associated with formation of amyloids in brain tissue, leading to (among others) Alzheimer’s disease and Creutzfeldt-Jakob disease [4,5,9,13].
When amyloidogenesis involves long polypeptide chains (unlike prions, which tend to be short), the end result is typically a mass of elongated fibrils which disrupt the functioning of otherwise healthy tissue [21-24].
From a molecular point of view, determining how proteins attain their intended tertiary conformations should go hand in hand with explaining the mechanisms behind the formation of misfolded structures [1,25].
When discussing the relation between residue sequences and the corresponding structural motifs we should acknowledge the fact that sometimes identical sequences produce different types of foldsparticularly helical or β-structural folds, depending on external conditions. Fragments which exhibit this property are referred to as chameleon sequences [26,27], and their existence is linked to the puzzling phenomenon of “intrinsically disordered” structures [28]. Several databases of chameleon sequences have been published [29,30] and they present an interesting study subject, given the involvement of chameleon sequences in prions and amyloids [31].
In-depth analysis of how helical folds may transform into β-structures points to the role of the so-called discordant helices. Such helices adopt α-conformations despite their notable sequential predisposition towards β-strands [31], as determined via numerical computations with GOR-IV and PSIPRED. This phenomenon can also be linked to the problem of chameleon sequences, enabling research into the mutual relations between conformational propensity, discordance, chameleons and amyloidogenecity [31]. Our work provides evidence for a causative link between discordant helices and chameleon sequences, and, consequently between discordant/ chameleonic protein segments and amyloidogenesis (especially with regard to prions).
Subsets of proteins listed in Gendoo and Harrison [31] have been subjected to analysis which focused on the stabilizing effects of an ordered hydrophobic core [32-34]. Assuming that the core is a major contributor to tertiary stabilization, we can attempt to determine its role in situations where the protein undergoes major structural rearrangement. This analysis bases on the so-called fuzzy oil drop paradigm, which models the “idealized” hydrophobic core with a 3D Gaussian superimposed onto the protein body. The function expresses hydrophobicity density throughout the protein body, peaking at its geometric center and reaching near-zero values close to the surface. Accordingly, hydrophobic residues are expected to be found deep inside the protein, while hydrophilic residues should be exposed on its surface. Real proteins conform to this model with variable accuracy: The actual (observed) distribution of hydrophobicity density depends on pairwise interactions as well as on the intrinsic hydrophobicity of each residue (see for example the hydrophobicity scales proposed [35-37]). Fragments which exhibit good accordance with the idealized model are thought to contribute to tertiary structural stabilization while fragments which diverge from the model may be thought of as inherently unstable (assuming that the molecule–as a whole -is stabilized by its hydrophobic core).
This work is the one in a set of publication which explain how the fuzzy oil drop model can be applied to interpret biological phenomena associated with proteins [38,39]. The model itself is thoroughly explained in Roterman et al. [38] here, we limit ourselves to a brief recapitulation of its basic tenets.
This work focuses on a set of proteins which include chameleon sequences listed in Gendoo and Harrison [31] as discordant helices and therefore susceptible to structural changes which might result in their classification as prions. Such sequences are responsible for post-translational modifications, leading to the formation of a β-sheet enriched conformer with altered biochemical properties.
Protein data sets
The analysis concerns a set of proteins identified in Gendoo and Harrison [31] as containing discordant helices and susceptible to amyloidogenesis. We limit our analysis to proteins which contain the VNITI sequence, so as to focus on prions with fewer than 100 residues. Larger proteins-especially multi domain ones-require a different interpretation and will be discussed in a separate paper. We have, however, extended our analysis with two proteins in which a different chameleon sequence (RYYEA) is present. While this addition does not significantly alter our study set, it nevertheless introduces an element of variability.
The fuzzy oil drop model
The fuzzy oil drop model asserts the existence of a so-called idealized distribution where the hydrophobicity density in a protein is modeled with a 3D Gaussian. Values of this function peak at the geometric center of the molecule and decrease along with distance from the center, reaching almost 0 on the surface (bell curve distribution). This idealized function is then confronted with the observed distribution which depends on the placement of each residue in the protein body, its interaction with neighbors as well as its intrinsic hydrophobicity (as given by a predetermined scale [35-37]). Comparison of both distributions reveals fragments where the theoretical (T) distribution closely matches observed (O) values (suspected of conferring structural stability), as well as discordant fragments which are probably inherently more flexible. Quantitative analysis of such local discordances may help identify sites prone to structural changes.
This paper discusses the applicability of fuzzy oil drop model to structure analysis of proteins. The detailed description of fuzzy oil drop model is presented in Roterman et al. [38]. The paper discussing the status of hydrophobic core in selected proteins representing the examples of similar sequence/different structure, different sequence/ similar structure is shown in Banach et al. [39].
To make the interpretation of the results easier the short description of the Kullback-Leibler divergence entropy [40] DKL calculation and interpretation is shown below.
In quantitative terms the degree of accordance (or, more accurately, the lack thereof) can be quantified using Kullback-Leibler divergence entropy criterion (DKL) [40]:
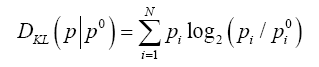
The value of DKL expresses the distance between the observed (p) and target (p0) distributions, the latter of which is given by the 3D Gaussian (T). The observed distribution (p) is referred to as O.
For the sake of simplicity, we introduce the following notation:
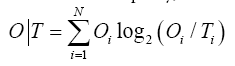
Since DKL is a measure of entropy it must be compared to a reference value. In order to facilitate meaningful comparisons, we have introduced another opposite boundary distribution (referred to as “uniform” or R) which corresponds to a situation where each effective atom possesses the same hydrophobicity density (1/N, where N is the number of residues in the chain). This distribution is deprived of any form of hydrophobicity concentration at any point in the protein body:
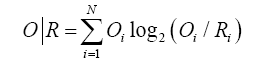
Comparing O|T and O|R tells us whether the given protein (O) more closely approximates the theoretical (T) or uniform (R) distribution. Proteins for which O|T>O|R are regarded as lacking a prominent hydrophobic core. To further simplify matters we introduced the following Relative Distance (RD) criterion:
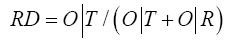
RD<0.5 is understood to indicate the presence of a hydrophobic core. Figure 1 presents a graphical representation of RD values, restricted (for simplicity) to a single dimension.

Figure 1: Graphical representation of fuzzy oil drop model hydrophobicity distributions obtained for a hypothetical protein reduced to a single dimension for simplicity. A-theorized Gaussian distribution (blue) while the chart C corresponds to the uniform distribution (green). Actually observed (red) hydrophobicity density distribution in the target protein B, while its corresponding value of RD (relative distance), and in D is marked on the horizontal axis with a red diamond. According to the fuzzy oil drop model this protein does not contain a well-defined hydrophobic core, because its RD value, equal to 0.408, is below the 0.5 threshold (or– generally–closer to T than R).
DKL (as well as O|T, O|R and RD) may be calculated for specific structural units (protein complex, single molecule, single chain, selected domain etc.) In such cases the bounding ellipsoid is restricted to the selected fragment of the protein. It is also possible to determine the status of polypeptide chain fragments within the context of a given ellipsoid. This procedure requires prior normalization of O|T and O|R values describing the analyzed fragment.
Applied tools
All protein structures were derived from PDB [41]. Some schematic diagrams were modified after PDBSum [42]. 3D visualizations were prepared with Pymol [43] and charts were plotted with Matplotlib [44].
Classification of residues involved in protein-protein interactions follows the PDBSum [42] standard for non-bonded contact map calculations (inter-atomic distance up to 3.9Å)
The presented analysis focuses on a set of proteins listed in Gendoo and Harrison [31]. Proteins containing the VNITI sequence have been divided into three groups:
1. Proteins, in which the VNITI sequence adopts a helical form: 1I4M-PrP Human [45], 1XYX-PrP Mouse [46], 1B10-PrP Golden Hamster [47], 2K56–Bank Vole [48], 3HAK PrP Human [49].
2. Proteins in which the VNITI sequence adopts a β-form: Immunoglobulin domains-1E4J [50], 1E4K [50], 1FNL [51]. All these proteins are extracellular domains of human Fc gamma RIII.
3. Proteins in which the VNITI sequence adopts a β-form: 1OP8–hydrolase E.C.3.4.21.78-Granzyme A (Hydrolysis of proteins, including fibronectin, type IV collagen and nucleolin. Preferential cleavage:
Arg-|-Xaa, Lys-|-Xaa>>Phe-|-Xaa in small molecule substrates) [52], 2VOV, 2VOW, 2VOX-metal-binding [53].
Introduction of two categories of chameleon sequences representing the β-form is aimed to distinguish the proteins of different biological activity.
Additionally the proteins containing the RYYEA sequence: Capsid- 1A6C (β-structural) [54] and a prion protein-1LG4 (helical) [55] were taken to include also other than VNITI sequence.
Group 1 proteins have been classified in Gendoo and Harrison [31] as prions containing discordant helices i.e., secondary folds which adopt a conformation which differs from their statistically computed natural predisposition. Discordant helices diverge from results obtained with GOR-IV and PSIPRED for the corresponding sequence for residues [31]. Similar properties are shared by 1LG4, which is also listed in Gendoo and Harrison [31].
VNITI sequence-Prp proteins with helical fragments (Group 1)
Proteins in which the VNITI sequence adopts a helical conformation are prions with about 100 aa. They provide a reference group with which their β-structural counterparts can be compared.
The above-mentioned proteins are an interesting study subject due to their pathogenic properties. Conformational changes which convert the Prion Protein (PrP) into its pathological form (PrP (Sc)) are regarded as the root cause behind devastating neurological conditions, such as spongiform encephalopathies.
Due to the high degree of structural similarity of prion proteins under consideration we have singled out the following structural elements: The N-terminal β-fragment (B1), the second β-fragment (B2) which combines with B1 to form a hairpin loop (B1+2), helical fragments jointly (HEL), the chameleon fragment (CHAM), the helical fragment which includes the chameleon sequence (FRAG), the entire domain (DOM) and a fragment comprising both β-folds in addition to the chameleon sequence (B1+B2+CHAM)-called COM. The rationale here is that the chameleon fragment interacts with the nearby β-hairpin to form a common layer (Figure 2).

Figure 2: 3D structure of a prion protein (3HAK), illustrating the close proximity between the ß-hairpin (yellow) and the helical chameleon sequence at 180- 184 (blue). This interaction justifies treating both subunits as a distinct unit (COM).
3HAK [49] is a protein consisting of 103 residues, with five helical fragments (including one short fragment with a single helical twist), in addition to two β-fragments which comprise an anti-parallel β-sheet (three residues per fold). The corresponding CATH classification is 1.10.790.10 Mainly Alpha Orthogonal Bundle. The chameleon fragment is present in the central part of one of the longer helices (residues 180-184). The protein includes a single disulfide bond, with one of its constituent Cys residues (number 179) located next to the chameleon sequence.
Presence of the C-terminal helix causes the 1I4M molecule as a whole to be non-globular [45]. Therefore all residues starting from T191 were excluded from calculations of the 3D Gaussian ellipsoid (which is important for the FOD model). As a result, this protein is described using two independent datasets.
RD values indicate that B1 (β-1), HEL (helix), CHAM (chameleon fragment) and FRAG (the entire secondary fragment containing chameleon sequence) are all accordant with theoretical expectations while the β-hairpin loop diverges from the model in most of the analyzed proteins. In light of these results, the chameleon sequence does not appear to stand out from among accordant fragments (except in 1I4M), despite its “distorted” status listed in Gendoo and Harrison [31].
In order to discern general trends, we have calculated RD correlation coefficients for all fragments and sets listed in Table 1. Of note is the high correlation between DOM and COM (0.807) and between DOM and CHAM (0.808), with the remaining coefficients also quite high (generally above 0.64). An interesting case is the status of B1 juxtaposed with the status of the entire domain and its constituent fragments–this is the only example of a negative correlation, which suggests that B1 counteracts the conformational propensities of other fragments even while remaining accordant with the FOD model. The B1/CAM, B1/ FRAG and B1/DOM correlation coefficients are -0.653, -0.757 and -0.748 respectively. Despite their limited statistical significance (small sample size), these values should not be disregarded.
| Protein | SS-bond | B1 | B2 | B1+2 | HEL | CHAM | FRAG | DOM | COM | N |
|---|---|---|---|---|---|---|---|---|---|---|
| 1I4M-a | 0.556 | 0.217 | 0.700 | 0.912 | 0.613 | 0.581 | 0.460 | 0.670 | 0.844 | 108 |
| 1I4M-a* | - | 0.399 | 0.564 | 0.856 | 0.389 | 0.376 | 0.352 | 0.576 | 0.683 | 71 |
| 1XYX-a | - | 0.378 | 0.189 | 0.633 | 0.355 | 0.138 | 0.320 | 0.440 | 0.511 | 112 |
| 1B10-a | - | 0.392 | 0.286 | 0.292 | 0.396 | 0.377 | 0.318 | 0.428 | 0.393 | 104 |
| 2K56-a | 0.385 | 0.437 | 0.324 | 0.515 | 0.390 | 0.365 | 0.322 | 0.497 | 0.428 | 113 |
| 3HAK-a | 0.409 | 0.402 | 0.026 | 0.762 | 0.427 | 0.222 | 0.415 | 0.454 | 0.666 | 103 |
Table 1: RD values calculated for the following fragments: fragment delimited by Cys residues forming a disulfide bond (SS-bond); first ß-fragment (B1), second ß-fragment (B2), ß-hairpin loop (B1+2), all helices taken together (HEL), chameleon sequence (CHAM), secondary fragment containing the chameleon sequence (FRAG), complete domain (DOM), aggregation of B1+2 with CHAM (COM). N indicates the number of residues in each chain. The asterisk (*) denotes the globular portion of the 1I4M chain (119-226) following elimination of its C-terminal fragment (119-190). Values listed in boldface satisfy RD>0.5.
The overall good accordance of the presented proteins with the theoretical model seems to suggest that there is no specific disordered fragment which could act as a “seed” for conformational changes leading to amyloidogenesis. To the contrary-all analyzed proteins seem to be highly soluble, with only the β-hairpin exhibiting “symptoms” of susceptibility to conformational changes by diverging from the FOD model (despite good accordance of its individual components).
Proteins with VNITI sequences adopting β-conformations-immunoglobulin domain-extracellular domain of human Fc gamma RIII (Group 2)
This group includes the following proteins:
1E4J-A–isolated extracellular domain of human Fc gamma RIII.
1E4K-C–extracellular domain of human Fc gamma RIII in complex with Fc domain-analysis focuses on chain C which corresponds to 1E4J-A.
1FNL–extracellular domain of human Fc gamma RIII.
The set of proteins where the chameleon (VNITI) sequence adopts a β-conformation comprises extracellular domains (FCγRIII) of low-affinity Fc gamma immunoglobulin. Domains under consideration include sandwich forms typical for immunoglobulin-like structures (CATH classification: 2.60.40.10). Each domain contains a single disulfide bond.
Comparing RD parameters for individual fragments (Table 2) indicates a very similar hydrophobic core status in each domain.
| 1E4K-C | 1E4J-A | 1FNL-A | |||
|---|---|---|---|---|---|
| FRAGMENT | RD | FRAGM | RD | FRAGM | RD |
| DOMAIN | 0.428 | DOMAIN (87-172) | 0.444 | DOMAIN | 0.441 |
| 87-91 BI | 0.492 | 87-91 BI | 0.419 | 90-94 BI | 0.347 |
| 95-99 BII | 0.261 | 95-97 BII | 0.339 | 98-102 BII | 0.301 |
| 103-110 BI | 0.504 | 103-110 BI | 0.368 | 106-113 BI | 0.344 |
| 113-115 H | 0.134 | - | - | - | - |
| 116-123 BII | 0.400 | 115-123 BII | 0.503 | 118-126 BII | 0.484 |
| 128-130 BII | 0.574 | 125-133 BII | 0.641 | 128-136 BII | 0.691 |
| 136-138 BI | 0.803 | 136-138 BI | 0.331 | - | - |
| 139-146 L | 0.423 | 142-146 H | 0.574 | 139-141 BI | 0.313 |
| 147-155 BII | 0.730 | 147-155 BII | 0.726 | 145-149 H | 0.580 |
| 158-162 BII | 0.175 | 158-162 BII | 0.240 | 161-165 BII | 0.190 |
| 165-170 BII | 0.538* | 165-170 BII | 0.518* | 168-173 BII | 0.468* |
| SHEET I | 0.530 | SHEET I | 0.399 | SHEET I | 0.328 |
| SHEET II | 0.344* | SHEET II | 0.504* | SHEET II | 0.475* |
| SHEET I+II | 0.451 | I+II | 0.508 | I+II | 0.475 |
| SS 107-151 | 0.378 | SS | 0.391 | SS 110-154 | 0.397 |
| CH 165-169 | 0.499 | CH 165-168 | 0.363 | CH 168-172 | 0.541 |
Table 2: RD values for immunoglobulins, together with the status of individual secondary folds, ß-sheets, fragments delimited by disulfide bonds and the chameleon sequence (CH). RD>0.5 (items listed in boldface) indicates poor agreement with the fuzzy oil drop model. B with Roman number-identification of ß-sheet.
Fragments listed in boldface diverge from the FOD model. Their similar location in all analyzed domains suggests that these domains follow a single stability/instability “blueprint” (as defined by the model), assuming that RD>0.5 indicates poor stability. Chameleon sequences are generally accordant with the idealized model and likely take part in structural stabilization of the molecule (the sole exception being 1FNL, where the RD value for the chameleon sequence exceeds 0.5; note, however, that very good conformance is observed if we elongate the analyzed fragment by 1 residue). Figure 3 depicts the location of chameleon sequences.

Figure 3: Schematic depiction of 1FNL domains (image borrowed from PDBSum [43]). “X” marks the chameleon sequence while the red and blue circles indicate proposed “latches”. The grey line corresponds to a signaling unit which directly links the N-terminal fragment and the loops (red line–residues interacting with antigen of the immunoglobulin) with the C-terminal fragment.
The availability of data concerning both the isolated domain (1E4J-A) and its complexed counterpart (1E4K-C) enables us to determine the influence of protein-protein interactions upon the structure of the hydrophobic core in domain 2 of 1E4K-C. Elimination of residues directly involved in interaction reduces the value of RD to 0.407 (from 0.428). This is a relatively small change but it nevertheless suggests that interaction with external proteins distorts the hydrophobic core.
Special attention should be directed towards the chameleon sequence, which appears related to this protein’s biological function. The sequence itself belongs to a structural motif frequently observed in immunoglobulins. Its N-terminal fragment adopts a β-conformation but it is clearly divided into two distinct sections, each of which belongs to a different sheet (referred to as the upper and lower core respectively Figure 3).
It is also worth noting that the C-terminal fragment is linked to the second fragment of the aforementioned β-fold (split into two parts). The link between the fragment at 17-21 and the C-terminal fragment creates a “latch”, bringing together the N-terminal fragment (typically in contact with the antigen) and the adjacent domain. It appears that the latch provides a means of transferring signals from the protein-binding domain to the domain which initiates the C1q complexation process, triggering the immune response. The ubiquitous nature of this structural motif in immunoglobulin suggests a common way of transmitting signals, regardless of their nature. This particular structure is well preserved, as shown in Table 3 which lists RD values for fragments of the latch and of the common β-system.
| DOMAINS | A–N-terminal | B–C-terminal | A+B |
|---|---|---|---|
| 1FNL–D1 | 0.426 | 0.660 | 0.616 |
| 1FNL–D2 | 0.300 | 0.468* | 0.335 |
| 1E4J-A–D1 | 0.366 | 0.564 | 0.514 |
| 1E4J-A–D2 | 0.272 | 0.518* | 0.314 |
| 1E4K-C–D1 | 0.564 | 0.616 | 0.639 |
| 1E4K-C–D2 | 0.261 | 0.538* | 0.344 |
Table 3: RD parameters computed for immunoglobulin domains present in our study set. The table lists values calculated for fragments forming the “latch” (cf. fragments A–N-terminal and B–C-terminal shown in Figure 3). Asterisks (*) indicate the presence of chameleon sequences. D1 and D2–identification of domains. The values given in bold distinguish the status discordant versus the expected one (RD>0.5).
The presented domains merit our attention due to the presence of chameleon sequences. If we accept the structural and functional interpretation of the “latch” (as described above), we should conclude that secondary conformation is not a goal unto itself but should instead be regarded as a step towards alignment to the global hydrophobic core-in the sense of the fuzzy oil drop model, which extends the definition of the “core” to include hydrophilic residues which shield the hydrophobic zone from contact with water. Thus, the conformation of the chameleon sequence is subordinate to global optimization of hydrophobicity density. It should also be noted that the actual (observed) hydrophobicity density distribution may deviate from the idealized model, and that such local deviations are often tightly coupled to the protein’s biological function, including potential deformations by which the protein fulfills its purpose. This specificity is readily evident in immunoglobulin domains discussed in [56-58].
Analysis of the status of the “latch” indicates that in domain 1 (17-21+82-87) its RD value is 0.616 while in domain 2 (168-172+91- 102) the corresponding value is only 0.493. This, in turn, suggests differentiation in terms of signaling mechanics. We are currently in the process of studying a large set of “latches”. Specific values obtained for proteins in our present study set are listed in Table 3.
Comparing the status of the “latch” in domains labeled “1” with the corresponding structure in domains labeled “2” reveals that only the former domains exhibit elasticity and mobility. This is likely related to the changes in environmental conditions at each stage of the signaling process.
Figure 4 illustrates the unification of the signal which seems to follow the logical AND protocol. The “latch” aggregates information regarding the presence of a complexation partner (155-158), transmitted through the 165-170 fragments, and another signal indicating N-terminal interaction (likely via the 95-99 fragments, which forms a mini-sheet with the 165-170 fragment).

Figure 4: Extracellular domain structure of human Fc gamma RIII (1FNL). Orange fragment–signaling unit (as shown in Figure 3); turquas band–corresponding unit in domain 1 (orange); yellow helix–artificially attached membrane component (not present in the crystal structure).
The above interpretation does not fully explain the role of the C chain in the 1E4J complex; however it is validated by the ubiquitous presence of the presented structural motif in immunoglobulin domains.
Proteins with VNITI sequences adopting β-conformations-hydrolase (1OP8) and metal-binding proteins (2VOV, 2VOW, 2VOX)
This group is distinguished by the chain length of its member proteins (longer than in the case of immunoglobulin domains), by their multidomain composition and their specific biological profile.
Hydrolase: Hydrolase is an enzyme (E.C.3.4.21.78) responsible for hydrolysis of proteins, including fibronectin, type IV collagen and nucleolin (preferential cleavage: Arg-|-Xaa, Lys-|-Xaa>>Phe-|-Xaa in small molecule substrate). Domain 1 (which includes the chameleon sequence) contains three catalytic residues. There are three disulfide bonds per domain. The crystal structure (1OP8) comprises six chains, enabling assessment of structures which consist of a large number of chains.
In all chains domain 1 is seen to include a well-ordered hydrophobic core, with very similar RD values. All secondary folds are also accordant with the idealized distribution (RD<0.5), with the exception of a single β-fold (Table 4 and Figure 5). The emergent β-structure (β-barrel) is likewise accordant, as is the chameleon sequence, which-in this case-adopts a β-conformation. Fragments delimited by SS bonds are accordant, which suggests that the hydrophobic core may play a role in guiding Cys residues towards their intended locations and ensuring the formation of disulfide bonds. This is in contrast to another frequently observed phenomenon, where SS-bonds introduce structural deformations which counteract hydrophobic effects and thereby stabilize the molecule in a specific locally unstable tertiary conformation [34].
| 1OP8-A | 1OP8-B | 1OP8-C | 1OP8-D | 1OP8-E | 1OP8-F | |
|---|---|---|---|---|---|---|
| CHAIN | 0.317 | 0.329 | 0.315 | 0.325 | 0.327 | 0.318 |
| 20-22 (B) | 0.430 | 0.481 | 0.492 | 0.444 | 0.480 | 0.441 |
| 134-140 (B) | 0.676 | 0.708 | 0.690 | 0.759 | 0.684 | 0.697 |
| 155-163 (B) | 0.372* | 0.450* | 0.432* | 0.404* | 0.434* | 0.417* |
| 164-169 (H) | 0.375 | 0.344 | 0.364 | 0.370 | 0.358 | 0.363 |
| 179-184 (B) | 0.258 | 0.224 | 0.203 | 0.204 | 0.157 | 0.189 |
| 197-202 (B) | 0.439 | 0.469 | 0.471 | 0.441 | 0.473 | 0.452 |
| 208-215 (B) | 0.471 | 0.488 | 0.519 | 0.483 | 0.490 | 0.483 |
| 226-231 (B) | 0.551 | 0.395 | 0.410 | 0.466 | 0.444 | 0.234 |
| β-sheet | 0.402 | 0.423 | 0.420 | 0.424 | 0.415 | 0.388 |
| 136-201 (SS) | 0.340 | 0.384 | 0.367 | 0.362 | 0.380 | 0.360 |
| 168-182 (SS) | 0.193 | 0.183 | 0.166 | 0.198 | 0.175 | 0.209 |
| 191-220 (SS) | 0.290 | 0.296 | 0.297 | 0.289 | 0.285 | 0.287 |
| CHAMELEON | 0.283 | 0.408 | 0.389 | 0.328 | 0.387 | 0.340 |
Table 4: RD values for hydrolase chains–full chain, individual secondary structure motifs (B–β-form; H–helix); β-sheet, fragments delimited by SS bonds and the chameleon sequence (158-162). Values listed in boldface correspond to RD>0.5.

Figure 5: Hydrophobicity density distribution profiles for each fragment listed in Table 4. Gray shading marks fragment containing the chameleon sequence.
Based on the location of fragments distinguished by fuzzy oil drop model as less stable (RD>0.5; inter-chain variability) and their relative proximity to catalytic residues, we may suspect their involvement in biological activity-by creating a locally unstable, flexible structure, capable of binding ligands. The placement of the chameleon sequence also indicates that it may participate in biological activity.
VNITI sequence in proteins binding metal ions
In Table 5 in Gendoo and Harrison [31] three proteins-1VOV, 1VOX and 1VOW-are listed as containing the VNITI chameleon sequence in its β-conformation. Each of these proteins binds a metal ion (Cu, Hg and Ca respectively) with help from kynurenine-a trypthophan metabolite [53].
| 2VOV-A | 2VOW-A | 2VOX-A | ||
|---|---|---|---|---|
| FRAGMENT | RD | RD | RD | |
| DOMAIN | 0.599 | 0.596 | 0.597 | |
| 49-55 BI | 0.517 | 0.546 | 0.528 | |
| 64-68 BII | 0.333 | 0.335 | 0.339 | |
| 92-100 BIII | 0.247 | 0.250 | 0.162 | |
| 105-109 H | 0.577 | 0.601 | 0.567 | |
| 110-112 BIV | 0.243 | 0.210 | 0.245 | |
| 119-124 H | 0.705 | 0.653 | 0.711 | |
| 126-128 BIV | 0.861 | 0.841 | 0.849 | |
| 129-132 BV | 0.379+ | 0.502 | 0.131+ | |
| 135-141 BI | 0.822 | 0.816 | 0.807 | |
| 145-154 BIII | 0.276 | 0.270 | 0.262 | |
| 155-158 H | 0.512 | 0.557 | 0.559 | |
| 164 – 172 BI | 0.455 | 0.460 | 0.454 | |
| 196-200 BII | 0.284 | 0.288 | 0.269 | |
| 203-205 BV | 0.273+ | 0.222 | 0.303+ | |
| 208-215 BI | 0.705 | 0.707 | 0.693 | |
| 238-251 BI | 0.582++ | 0.527++ | 0.513++ | |
| 259-261 BVI | 0.149 | 0.154 | 0.161 | |
| 268-269 H | 0.298 | 0.221 | 0.203 | |
| 270-272 BI | 0.118 | 0.119 | 0.085 | |
| 278–286 BIII | 0.470++ | 0.478++ | 0.471++ | |
| 290-299 BI | 0.765 | 0.751 | 0.760 | |
| 300-302 BVI | 0.704 | 0.704 | 0.700 | |
| 304-308 BVII | 0.600 | 0.604 | 0.560 | |
| 312-316 BVII | 0.511 | 0.619 | 0.503 | |
| 325-334 BIII | 0.415* | 0.421* | 0.624*+++ | |
| β-sheet I | 0.624 | 0.621 | 0.613 | |
| II | 0.296 | 0.301 | 0.288 | |
| III | 0.346 | 0.349 | 0.330 | |
| IV | 0.399 | 0.391 | 0.409 | |
| V | 0.444 | 0.448 | 0.552 | |
| VI | 0.466 | 0.458 | 0.457 | |
| VII | 0.553 | 0.619 | 0.530 | |
| CHAMELEON | 0.551 | 0.529 | 0.549 | |
Table 5: RD values calculated for domains, secondary folds, β-sheets and the chameleon (329-333) sequence in metal-binding proteins (2VOV, 2VOW and 2VOX). Asterisks mark fragments which contain the chameleon sequence. “+” marks denote ion complexation sites: “+” corresponds to Cu2+, “++” corresponds to Ca2+ while “+++” corresponds to Hg ions.
The chameleon sequence is located in the C-terminal fragment of the chain whose total length is 290 aa. It belongs to a β-fold which, in turn, participates in a β-sheet composed of four fragments (one of seven β-fragments present in protein). The four components of the β-sheet form a quasi-barrel, although it is not identified as such under CATH criteria.
The group comprises the following metal-binding proteins: 2VOV– Cu2+ and Ca2+; 2VOW–Ca2+ and 2VOX–Ca2+, Cu2+ and Hg2+.
Each chain contains 290 residues, forming a single domain, with no discernible hydrophobic core (as evidenced by FOD calculations). The system of β-folds creates a quasi- β-barrel (not identified as such under CATH criteria), with seven β-sheets of which the first and the seventh diverge from the hydrophobic core model. Similarly to the whole structure, the chameleon sequence does not adhere to the theoretical model (Figure 6).

Figure 6: 3D visualization of the structure of 1OP8. Red band–134-140 fragment (discordant and therefore possibly inherently unstable); blue band–chameleon sequence; yellow band–fragment at 226-231 whose status varies between chains comprising the protein’s crystal structure. Orange space filling–enzymatic residues.
In the case of 2VOX, the chameleon sequence is directly involved in complexation of mercury ions, as indicated by its hydrophobicity density profile (Figures 7 and 8).

Figure 7: RD profiles for selected fragments listed in Table 5. Gray background marks the location of chameleon fragment. Vertical lines denote fragments that host metal ion complexation sites.

Figure 8: 3D visualization of 2VOV, with residues involved in metal ion complexation marked in yellow. The red band corresponds to the chameleon sequence.
The β-conformation of the chameleon sequence observed in 2VOV, 2VOW and 2VOX seems related to structural stabilization. The FOD status of the sequence does not differ from that of the molecule, which suggests that the sequence is aligned with the structure of the protein as a whole.
RYYEA chameleon fragment
We have decided to augment our discussion of VNITI sequences with analysis of a different chameleon sequence–RYYEA. Our aim is to show that the status of the sequence is subordinate to the overall conformation of the molecule rather than to local conditions.
Our analysis covers two proteins: a capsid protein (1A6C) and a prion protein (1LG4). According to Gendoo and Harrison [31] both include a discordant chameleon sequence, yet they differ greatly with regard to their chain length-which is an important criterion under the fuzzy oil drop model. In the capsid protein the β-structural chameleon sequence forms part of a longer β-fragment which itself participates in a four-part β-sheet (CATH 2.60.120.20-mainly β-sandwich; although it should be noted that the relative abundance of β-sheets-7 in total-makes the sandwich form quite complex). Two domain-like subunits can be distinguished, and while these subunits share a large subset of chain fragments, domain decomposition is important for the FOD model. Consequently, our analysis of this protein’s hydrophobic core may not be fully objective. The domain as a whole diverges from the model (Table 6), as does the β-sheet which includes the chameleon sequence, although the sequence itself remains accordant with theoretical expectations.
| 1LG4 | 1A6C | ||||
| FRAGMENT | RD | FRAGMENT | RD | ||
| DOMAIN | 0.482 | DOMAIN | - | 0.731 | |
| Helices | 71-82 | 0.414* | β-sheet I | - | 0.674 |
| 83-85 | 0.700 | II | - | 0.550 | |
| 100-116 | 0.337 | III | - | 0.800 | |
| 125-142 | 0.381 | IV | - | 0.696 | |
| - | - | V | - | 0.617 | |
| SS-bond | 94-145 | 0.520 | VI | - | 0.328 |
| 108-140 | 0.383 | VII | - | 0.767 | |
| CHAM | 76-80 | 0.292 | CAM. | 193-197 | 0.409 |
| - | - | FRAGM | 189-200 | 0.623 | |
Table 6: Comparison of RD values for the 1LG4 prion protein and the 1A6C capsid protein. In 1LG4 separate values are listed for each secondary fold while in 1A6C we focus only on individual β-sheets. FRAGM corresponds to the fragment which includes the chameleon sequence in 1A6C.
The prion protein (1LG4) is a single-domain protein comprising 99 residues, with the helical sequence adopting a helical conformation. The protein includes two disulfide bonds which are, however, distant from the chameleon sequence and do not appear to directly influence its conformation. The protein as a whole conforms to the hydrophobic core model, as does the chameleon sequence. The only fold identified as discordant is a fragment of the loose loop at 83-85 (irregular helix) (Figure 9).

Figure 9: 3D visualization of the chameleon sequences in 1A6C and 1LG4 (red). Pairs of Cys residues forming SS bonds are marked in orange and yellow respectively.
Even though comparative analysis of such dissimilar proteins must necessarily be generalized, we should note that in most cases the status of the chameleon sequence corresponds to that of the entire protein (the only exception is 1A6C). 1A6C exhibits a fairly disordered distribution of hydrophobicity density. The protein attains structural stability by forming a multimolecular capsid-type aggregate. This, however, requires a specific hydrophobicity density distribution, approximating that of a membrane, with water present on either side of a planar surface (Figure 9).
Analysis of RD values listed in Table 6 again suggests that chameleon sequences tend to align themselves structurally with the protein molecule, contributing to the formation of a stabilizing hydrophobic core. The fuzzy oil drop model reveals the subtle interplay between the status of chameleon sequences and that of other fragments comprising the protein chain.
The prion proteins analyzed in this work appear to contain prominent hydrophobic cores. This property should result as good solubility–which is somewhat surprising given that prions exhibit a notable tendency to cluster and generate fibrillary multimolecular structures commonly referred to as amyloids. This conclusion is based on the specificity of fuzzy oil drop model assuming the hydrophilic shell covering the entire surface of the protein (without local disorder in the context of fuzzy oil drop model). The hydrophilic surface ensures accordance with the surrounded water environment (the solubility is the result of this status). High accordance of the hydrophobic core structure with the idealized one is in contrast to expected elasticity, which may occur (taking the fuzzy oil drop criteria), when the high entropic contact of hydrophobic residues exposed on the surface is observed or when the local deficiency of hydrophobicity may push other fragments of polypeptide chain to fit to the cavity of lower than expected hydrophobicity level.
The authors of Gendoo and Harrison [31] attribute the presence of the so-called “distorted helices” in prions (with low affinity for this type of fold) as a promoter of conformational changes resulting in the formation of complexes, including amyloids. However, no such predisposition is evident in our analysis. While assessing the role of chameleon sequences we have identified fragments which remain highly accordant with the FOD model, eliminating those which do not conform to the model with high accuracy. With regard to prion proteins, the chameleon sequence always survives such pruning, suggesting its involvement in the formation of a protein-wide hydrophobic core.
To-date experience with the fuzzy oil drop model indicates that a domain consisting of 100-150 residues, folding on its own, will typically produce a structure which is highly consistent with the theoretical model. Local deviations from the idealized distribution are usually associated with protein complexation sites (excess hydrophobicity on the surface) or ligand binding cavities (local hydrophobicity deficiencies).
As shown in Roterman et al. and Banach et al. [38,39], there is no evidence of direct involvement of chameleon sequences in biological activity in any of the analyzed proteins. Based on their observed alignment with the hydrophobic core structure, we can conclude that such sequences typically adopt the structural form determined by the structure of hydrophobic core independently on the local stabilization/ unstabilization of particular chain fragment.
In summarizing the presented work, as well as the two papers in the series [38,39], we should point to the fact that the conformation of chameleon sequences generally remains in agreement with the overall shape of the protein’s hydrophobic core, including its encapsulating hydrophilic shell. Secondary structural characteristics result from alignment with the idealized hydrophobicity density distribution profile (or any local deviations from this profile). A hypothetical protein in which the observed distribution is a perfect match for the 3D Gaussian would be perfectly soluble but incapable of specific interactions with any other molecules–whether proteins or ligands. It seems that local irregularities, well preserved across protein families, are a critical factor in ensuring biological specificity. This property also affects the immediate neighborhood of the active group, where sufficient flexibility is required in order to accommodate a ligand or complexation partner–as evidenced by immunoglobulin-like domains (particularly 7FAB and 1SBT [39]).
The concept of structural transformations leading to the formation of amyloid fibrils treats structural changes as a form of cooperative rearrangement affecting the entire molecule. The fuzzy oil drop model lends itself well to tracking and describing such changes.
The cooperative character of conformational changes may follow from changes in the protein’s external environment which, under certain conditions, favors alternative conformations. The protein itself merely adapts to the altered external force field. We thus postulate simulating the folding process in an external field which differs from the 3D Gaussian [59]. The influence of external forces upon the protein’s biological properties stems from the close relation between tertiary conformation and the aqueous environment. Consequently, the formation of amyloid structures–for example via prolonged shaking-can be interpreted (on the grounds of the FOD model) as caused by yet-undetermined changes in the structural properties of water [60,61]. In light of this observation, effects which promote in vitro amyloidogenesis may act upon the water environment rather than on the protein itself-with the protein simply adapting to external changes (as stipulated by the fuzzy oil drop model). The amyloid, having been created under altered conditions, is not influenced by factors which require “normal” conditions (especially with regard to the structural properties of water)-such as proteolytic enzymes. A specific property of amyloids is their ability to bind Congo red. This phenomenon suggests a way to generate the structure of the amyloid protein on the basis of compatibility with the dye, depending on whether it is complexed in its monomeric [62] or supramolecular [63] form. The status of complexes and the status of the interface in protein-protein complexes described using fuzzy oil drop model reveals different category of interfaces [64]. This observation seems to be useful for amyloidogenesis analysis, the plausible hypothesis of which is shown in Roterman et al. [38].
The Authors are grateful to Piotr Nowakowski and Anna Śmietańska for technical aid. This work was funded by Collegium Medicum grant no. K/ZDS/006363 under the KNOW system.