Journal of Thermodynamics & Catalysis
Open Access
ISSN: 2157-7544
ISSN: 2157-7544
Research Article - (2016) Volume 7, Issue 4
The Cu2+ or Fe3+ substituted Cs2HPMo12O40 was synthesized to study the effect of transitional metal cations on selective catalytic oxidation of methacrolein by polyoxometalates. Adding of Cu2+ could reduce acidity and enhance oxidizing ability of the catalyst. The catalytic experiments showed that the addition small amount of Cu2+ to the catalyst could increase the conversion of methacrolein from 65% to 96% and the selectivity to methacrylic acid to 55%, at 300-340°C. It is found adding of Cu2+ could promote the reduction process of catalysts should be the main reason of the performance enhancement. Adding of Fe3+ to the catalyst could induce some new weak acidic sites due to the formation of iron aqua complexes. Whereas, Fe3+ had no significant effect on redox ability of the catalyst. The conversion of methacrolein could reach 96% with the existence of Fe3+. However, the addition of Fe3+ had no significant effect on selectivity to methacrylic acid.
Keywords: Heteropoly catalysts, Support, Selective oxidation, Methacrolein, Methacrylic acid
Heteropoly catalysts have attracted a lot of attention in oxidation, acid and shape-selective catalysis [1-5]. The oxidation of methacrolein (MAL) to methacrylic acid (MAA) is a successful example of industrial applications of heteropoly catalysts [6-8]. Due to the high cost and low turnover frequency of this kind of catalysts, researches on supported heteropoly catalysts for higher catalytic efficiency are always attractive.
Silica is a kind of cheap, useful, and thermal stable supports, and some heteropoly acids have been supported on silica [9-19]. Kanno et al. found that SiO2 supported H4PMo11VO40 (HPAV) had good catalytic activity for oxidation of MAL to MAA, especially when using aminated SiO2 as the support, on which the TOF was enhanced 5 times. Thermal stability of this kind of supported heteropoly acids was poor as HPAV was decomposed to MoO3 and V2O5 [15,16]. Similarly, the SiO2 supported H3PMo12O40, reported by Gomez-Sainero, showed low catalytic activity due to the loss of Brönsted acids sites, and H3PMo12O40 was decomposed to MoO3. To solve this problem, ZrO2 was added as a cocatalyst, and catalytic activity was obviously improved [12]. H4SiW12O40 and H3PW12O40 were supported on porous SiO2. However, these two heteropoly acids reacted with silica and formed [=SiOH2]3PW12O40 and [=SiOH2]xH4-xSiW12O40, which leaded to the decomposition of the Keggin structure due to the existence of over acidified protons [13]. Ballarini prepared heteropoly acids in SiO2 by co-gelation procedure. The catalysts showed poor thermal stability due to the replacement of P by Si as center atoms [14]. Silica-based ordered mesoporous molecule sieves, such as SBA-15, MCM-41 and KIT-6 were also utilized as support for heteropoly acids. These supported catalysts showed better catalytic activity than the bulk ones. However, the supported heteropoly acids have the same drawbacks as those catalysts supported on SiO2, e.g., low thermal stability [9,10]. So it is necessary to find a way to modify silica and improve the thermal stability of supported heteropoly catalysts. The addition of counter ions with large volumes, such as K+, Cs+, NH4, is an effective way to improve thermal stability of heteropoly compounds [20]. The addition of counter ions, such as Cs+, also can improve the catalytic activity for the oxidation of MAL to MAA and increase the selectivity for MAA [21,22]. Therefore, suitable counter ions are needed to prepare thermal stable supported heteropoly catalysts. In this study, four different silica-based supports (i.e., porous SiO2 SBA-15, KIT-6 and HY zeolite) with different pore sizes were used as the supports to load H4PMo11VO40, and Cs+ was added as the counter ion. The preparation condition, counter ion content and loading amount were optimized to get thermally stable supported heteropoly catalysts. The decomposition mechanism of the supported heteropoly catalysts during calcination and oxidation of MAL to MAA were studied in detail.
Preparation of catalysts
H4PMo11VO40 (HPAV) was prepared from MoO3 (Sinopharm Chemical Reagent Co., Ltd.), V2O5 (Tianjin Guangfu Fine Chemical Research Institute) and 85% phosphoric acid (Sinopharm Chemical Reagent Co., Ltd.) according to published procedures [3]. Briefly, 5 g MoO3 and 0.287 g V2O5 were added to 100 mL deionized water, which was subsequently heated to reflux. Then, 0.364 g of 85% phosphoric acid was added to the mixture, which was refluxed with vigorous stirring for 5 h. A deep-orange solution then formed. The insoluble solid was filtered, and the solution was evaporated at 80°C, obtaining a bright orange solid. Porous SiO2 was purchased from Sinopharm Chemical Reagent Co. Ltd., SBA-15, KIT-6 and HY zeolite were prepared following the published procedures [23-25], and the preparation processes were described in Supporting Information. The structures of the obtained supports were confirmed by FT-IR, Raman, BET, XRD and SEM. The supported HPAV was prepared by the following procedure. 1 g HPAV was dissolved in 10 mL deionized water, then 1 g supports (porous SiO2, SBA-15, KIT-6 or Y zeolite) were added to the HPAV solution, and the mixture was vibrated at 40°C for 12 h. After the vibration, the mixture was dried in an oven at 80°C for 12 h. The yellow powder was the prepared supported HPAV. Supported CsH3PMo11VO40 was prepared by the following way. 0.11 g CsNO3 was dissolved into 10 mL deionized water, and 1 g SBA-15 was added to the solution, then the mixture was vibrated at 40°C for 12 h. After then, the mixture was dried in oven at 80°C for 12 h and SBA-15 with CsNO3 was prepared. 1 g HPAV was dissolved in 10 mL deionized water, and it was dripped into SBA-15 with CsNO3, then the mixture was vibrated at 40°C for 12 h. After the vibration, the mixture was dried in oven at 80°C for 12 h. The bright yellow powder was the prepared catalysts containing Cs. CsH3PMo11VO40 supported on other supports were prepared following the similar processes and were named as CsPAV/SiO2, CsPAV/SBA, CsPAV/KIT and CsPAV/Y. Catalysts with different supporting amounts were named like this, 0.5CsPAV/SBA, which means 0.5 times of SBA-15 weight of catalysts was supported on SBA-15. All catalysts were calcined at 360°C in a flow of air at a rate of 80 mL·min-1.
Characterization of the catalysts
Fourier transform infrared (FT-IR) spectroscopy was performed on an FT-IR spectrometer (Nicolet 380, Thermal Electron Corporation) with anhydrous KBr as standard. Raman spectroscopy was conducted by a laser Raman spectrometer (HR800 UV, Horiba Scientific Co. Ltd) with a 100 mV laser, equipped with a wavelength of 514 nm and CCD detector. Ultraviolet-visible (UV-vis) diffuse reflectance spectroscopy was recorded on a UV-vis spectrophotometer (UV-2550, Shimadzu Corporation). Powder X-ray diffraction (XRD) was performed on an X-ray diffractometer (Bruker D8 Advance X-ray powder diffractometer). Samples (about 10 mg) were analyzed by conducting thermogravimetry/differential thermal (TG/DTA, analysis using a simulaneous DTA-TG apparatus DTG-60H, Shimadzu, Japan), which allowed both TGA and DSC curves to be recorded simultaneously. The samples were heated from room temperature to 650°C at heating rate of 5°C/min. The oxidation and acid properties of catalysts were tested by hydrogen temperature programmed reduction and ammonia temperature programmed desorption on AutoChem II Chemisorption Analyzer (Micromeritics). The microcosmic pattern of the catalysts was observed by scanning electron microscope (SEM, SU8020, Hitachi Electronic Electric Appliance Company, Japan) and field emission electron microscope (TEM, JEM-2100F, JEOL, Japan). The X-ray photoelectron spectroscopy (XPS) spectra were recorded by a Kratos Axis Ultra DLD spectrometer with monochromatic Al Kα radiation. The surface area and pore structure of the catalysts were tested by Surface area and Pore Analyzer (Mocromeritics ASAP 2460, US). 29Si NMR and 31P NMR were performed on Bruker Avance III (400M).
Determination of catalytic activity
Catalytic oxidation of MAL was performed in a fix bed reactor under 310°C at atmosphere pressure. A total of 0.6 mL catalyst was loaded into the fix bed reactor and the contact time was about 2 s. Before the detection, temperature was increased to 310°C, then a reactant mixture of MAL (4.4 vol.%), O2(11.1 vol.%), H2O (17.8 vol.%) and N2 (balance) was fed into the reactor to start the reaction. The reaction products were collected and analyzed using GC (SP-7890, Lunan Ruihong Chemical Engineering Instrument Co. Ltd) equipped with a flame ionization detector and a TDX-01 packed column. The catalytic activity of the catalysts was evaluated by TOF, which was calculated using Formula 1:
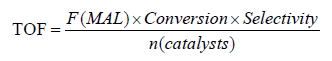 Formula 1
Formula 1
where F(MAL) was the molar flow rate of methacrolein (MAL) (mol·min-1), Conversion was the conversion of MAL, Selectivity was the selectivity of methacrylic acid (MAA) and n (Catalysts) was the mole amount of heteropoly catalysts (the supports was not included).
Characterization of the catalysts
To illustrate the changes of structure and chemical properties of the catalysts during calcination and reaction, FT-IR, Raman, XRD, BET, TG-DTA, XPS, 29Si NMR and 31P NMR were performed..
FT-IR: Figure 1 showed the FT-IR spectra of uncalcined, calcined and used CsPAV and supported catalysts with different supporting amounts. The four characteristic vibration bands (1062-1065 cm-1, vs(P-O); 963-966 cm-1, vs(M=O); 865-872 cm-1, vs(Mo-Ob-Mo); 777- 797 cm-1, vs(Mo-Oc-Mo) of heteropoly compounds were shown in these spectra [26-28]. The three different vibration bands [1080, 812 and 470 cm--1 vs(Si-O)] of supports were also present in the spectra [29]. Compare with the bulk CsPAV, the vibration bands of Keggin structure in the supported catalysts shifted to high wavenumber due to the interaction between CsPAV and supports. The weak vibration appeared around 1000 cm-1 may be attributed to Si-O in Keggin structure which was caused by the replacement of P by Si [14]. To prove this replacement, XPS, 29Si and 31P NMR were used to characterize the catalysts, which would be discussed in section 3.1.8. The spectra showed that CsPAV was well supported on the supports. A new band appeared at 1034 cm-1 in the spectra of the calcined CsPAV corresponding to V-O caused by the migration of V from Keggin structure to the secondary structure of catalysts [8]. And no other obvious changes can be found in these FT-IR spectra of the supported catalysts. It demonstrates that Keggin structure was well maintained after calcination. No obvious changes were found in the spectra of the used catalysts. The catalysts with different loadings showed almost the same spectra before calcination, after calcination and reaction. However, there were also some differences between the spectra of catalysts with different loadings. With the increase of loading amount, the vibration bands of Keggin structure shifted to low wavenumber (1081 to 1062 cm-1, vs(P-O); 965 to 963 cm-1, vs(M=O); 868 to 865 cm-1, vs(Mo-Ob-Mo)); 797 to 781 cm-1, vs(Mo-Oc-Mo) because of the decrease of interaction intensity between HPAV and support. To enhance thermal stability of the supported catalysts, Cs contents and loading amount were tuned. In the spectra of HPAV/SBA (Figure S1, Supporting Information), some vibration bands (994 -1, vsMo=O; 589 cm-1, vsMo-O-Mo) belonging to MoO3 were shown. It suggests that HPAV was decomposed during the preparation process. HPAV might react with silica species, which led to the decomposition of the catalysts [15]. After calcination and reaction, another vibration band of MoO3 was shown at 869 cm-1, and the other two bands became stronger, which suggests the further decomposition of the catalysts. For catalysts with Cs, only a weak vibration at 589 cm-1 was observed. After calcination and reaction, no other changes could be detected by the FT-IR, indicating that the addition of Cs could improve thermal stability of supported catalysts.

Figure 1: FT-IR spectra of uncalcined, calcined and used supported catalysts with different supporting amount (a) FT-IR spectra of uncalcined catalysts; (b) FT-IR spectra of catalysts which were calcined at 350°C for 12 h; (c) FT-IR spectra of catalysts which have been used in reaction.
Raman: In Figure 2, the typical main vibration bands (625 cm-1, Mo-Oc-Mo; 876-909 cm-1, Mo-Ob-Mo; 984 cm-1, Mo-O-P; 1002 cm-1, Mo=Od; 230-250 cm-1, the deformation vibration of the terminal M=O groups and the entire framework of Keggin structure.) of the catalysts with Keggin structure were shown [8,30]. The vibration band appearing at 1120 cm-1 could be assigned to the Si-O band in the supports. After calcination, the vibration bands at 250 and 984 cm-1 in the spectra of 0.5CsPAV/SBA and 1CsPAV/SBA transformed from a shoulder to a single band because of loss of crystalline water and partial decomposition of Keggin structure during calcination [30]. For other catalysts, vibration bands at 230-250 and 825 cm-1, which belong to MoO3, were shown, indicating that the supported catalysts were decomposed significantly during calcination. Decomposition of the catalysts was not observed as the vibration bands of MoO3 didn't appear in the spectra of 0.5CsPAV/ SBA and 1CsPAV/SBA, while the vibration bands of MoO3 in the spectra of 2CsPAV/SBA and 3CsPAV/SBA indicated decomposition of these catalysts. Integral Keggin structures were detected in the spectra of 4CsPAV/SBA and 5CsPAV/SBA. It indicates that more supported CsPAV was decomposed in the catalysts with more loading amount than the ones with less loadings. The vibration band of Si-O became too strong after calcination because of the interactions between catalysts and supports during calcination. After reaction, the spectra of CsPAV remained the same. While the spectra of the support became very strong, and almost no vibration bands of Keggin structure were detected. The vibration bands emerged at 825 and 1002 cm-1 can be assigned to Mo-O in MoO3 in the spectra of used 2CsPAV/SBA and 3CsPAV/SBA. It seems that the supported catalysts were almost completely decomposed after reaction. The vibration bands of MoO3 was supposed to be detected in the spectra of used 0.5CsPAV/SBA and 1CsPAV/SBA, yet only strong vibration bands of supports were detected. The vibration bands of the decomposed catalysts might be clouded by the vibration bands of supports. Vibration bands, which belong to Keggin structure can be found in the spectra of 4CsPAV/SBA and 5CsPAV/SBA, which indicates that with the increase of the loading amount, the surface of the catalysts would not interact with the support directly and decomposition can be avoided.

Figure 2: Raman spectra of uncalcined, calcined and used supported catalysts with different supporting amounts
XRD: XRD patterns of the catalysts (2θ=10.6°, 18.4°, 21.2°, 23.8°, 26°, 30.2°, 32.2°, 35.6°, 38.9°, 43.2°, 47.4°, 51.2°, 55°, 61.8°) showed the typical cubic crystalline of cesium molybdovanadophosphoric hydrate (Csh3PMo11VO40·nH2O) (Figure 3). The peak at 8° could be assigned to HPAV. No peaks related to MoO3 or H4PMo11VO40 were found in the patterns of the catalysts before calcination, suggesting that HPAV did not completely react with Cs+ to form CsPAV. A new peak emerged at 27.3° in the patterns of calcined supported catalysts which could be assigned to MoO3. It indicates that the supported catalysts were partially decomposed after calcination. The peak at 8° belonging to HPAV disappeared because of the decomposition of HPAV. The diameters of crystal were calculated by Scherrer formula, Dhkl=kλ/βcosθhkl (where k is shape factor, cubic is 0.9; λ is the wavelength of X-ray (nm); β is FWHM (radian); θhkl is diffraction angle (radian).). The diameters of the catalysts increased with the increase of loading amount of CsPAV (20, 21.5, 22, 39, 43.9 and 44.7 nm, respectively), which indicates that CsPAV crystal grew larger and lager with the increase of loading amount of CsPAV. With the loading amount increasing to 2, MoO3 patterns emerged at 10°, and peaks of MoO3 became stronger and stronger with the increase of loading amount of CsPAV. It indicates that Keggin structure was decomposed during the preparation process, and more MoO3 formed with the increase of loading amount. After calcination and reaction, the patterns of MoO3 became more obvious, which showed that catalysts were further decomposed during the calcination and reactions. No obvious changes in the patterns of catalysts were observed after reaction. The sizes of the supported catalysts changed little after calcination and catalytic reaction, suggesting that most of Keggin structures kept integral after reaction. The XRD patterns of HPAV/SBA were obviously different from CsxPAV/SBA (Figure S2). Some peaks appeared at 10.5°, 19.1°, 25° and 28.9°, which showed that HPAV were well dispersed on SBA-15. The wide peaks showed that the diameter of HPAV was very small. HPAV might disperse on SBA-15 by a single molecule, because the surface area of SBA-15 used in this experiment was about 1000 m2/g, while the diameter of a single HPAV molecule is about 1 nm [15,16], and HPAV/SBA was 1 g HPAV supported on 1 g SBA-15, while 4 g HPAV could form a single molecule layer on 1 g SBA-15. The patterns of the catalysts with Cs are similar. The main peaks could be assigned to CsPAV, and the peaks of HPAV (8°) were also shown in the patterns of CsPAV/SBA and Cs2PAV/SBA. And it disappeared in the patterns of Cs3PAV/SBA, which demonstrated that all PMo11VO4- reacted with Cs+ during preparation of Cs3PAV/SBA. The sizes of Cs1~3PAV crystals on the support are almost the same (21-22 nm). The supported HPAV was decomposed after calcination, so the peaks of MoO3 were obviously observed. HPAV in CsxPAV/SBA was also decomposed during calcination. The peaks at 23.5° and 24.9° show that some integral Keggin structures remained after calcination. The peak of MoO3 (27.3°) was also found in the patterns of CsPAV/SBA and Cs2PAV/SBA. No peaks of MoO3 were found in Cs3PAV/SBA. So the appearance of MoO3 was due to the decomposition of HPAV in the supported catalysts, indicating that increasing of Cs content can improve thermal stability of the catalysts.

Figure 3: XRD patterns of uncalcined, calcined and used supported catalysts with different supporting amounts.
SEM and TEM: SEM and TEM could reveal the state of the supported catalysts. Figure 4 shows that the bulk CsPAV has no particular shape and size. The images of supported catalysts showed that all the supports kept integral after preparation and calcination. CsPAV was clustered on the surface of the supports in the sizes of 30-150 nm which were in accordance with the calculated results of XRD. Although the supports were different, CsPAV showed the same shape. The CsPAV might form from the following process. Firstly, CsNO3 was uniformly dispersed on the supports. After the HPAV solution dripped onto the supports with CsNO3, some Cs+ was added into the solution, which could reacted with PMo11VO40, forming CsPAV on the outer surface of the supports. Some PMo11VO40 might come into the pore of the supports and react with Cs+ to form CsPAV on the pore wall. With the increase of loading amount, CsPAV particles on the surface of SBA-15 increased obviously. When the loading amount was increased to 75%, almost all the holes in SBA-15 were filled by CsPAV. With the loading amount further increasing, the surface of SBA-15 was all covered by CsPAV. The CsPAV particles also clustered by themselves. No independent HPAV particles could be found in HPAV/SBA (Figure S3), indicating that HPAV was well dispersed on the SBA-15. TEM images shows that CsPAV particles were supported on the surface of the carriers. Some CsPAV might enter the pore of SBA-15. TEM images also show that CsPAV cumulated in the entrance of the channels.

Figure 4: SEM (Top) and TEM (Bottom) images of CsPAV/SBA.
N2-sorption: Figure 5 showed that the surface area and pore diameter of SBA-15 were about 1017.8 m2·g-1 and 6.6 nm, respectively. The nitrogen absorption isothermal lines of SBA-15 show that it was a kind of mesporous material with some micropores which were indicated in the part of nitrogen absorption isothermal lines below 0.1. After CsPAV being loaded on SB A-15, the surface area, pore volume and the ratio of micropores decreased with the increase of loading amount of CsPAV. The surface area decreased from 1017.8 to 70.0 m2·g-1. As mentioned above, some CsPAV entered the pores of SBA-15. The average diameter of CsPAV was higher than 20 nm. So the decrease of surface area was due to the clog of CsPAV in the entrances and channels of pores. When the supporting amount increased to 3, the micropores almost disappeared and some new pores with average diameter of 3.8 nm were formed. As can be seen in the Nitrogen absorption isothermal lines and pore width distribution of CsPAV, its pores were accumulation ones with wide diameter distribution. So these pores with average diameter of 3.8 nm were formed when the CsPAV molecules entered the pores of supports. With the further increase of loading amount, the ratio of pores with average diameter of 3.8 nm increased obviously, and the hysteresis loops of 4HPAV/SBA and 5HPAV/SBA were divided into two parts. It suggests that with the increase of loading amountof CsPAV, more catalysts entered the pores of supports. Some pores with wide diameter distribution were also formed in 4HPAV/SBA and 5HPAV/SBA which indicates the accumulation of CsPAV crystals on the outer surface of the supports.

Figure 5: Nitrogen absorption isothermal lines and pore width distribution of supported catalysts with different supporting amount.
TG-DTA: TG-DTA was performed to figure out the changes during calcination and thermal stability of the supported catalysts. The TG curves of SBA-15 showed a two-stage loss (Figure 6). The first one between 100 and 150°C can be assigned to loss of absorbed water, which indicated that about 7 wt% waters existed in SBA-15. The second stage could be attributed to the loss of crystalline water. The TG curves of HPAV/SBA exhibited the first one at 150-200°C, which was higher than that of SBA-15. The reason was that besides the absorbed water in SBA-15 and HPAV, there also existed crystalline water in HPAV which needed higher temperature to be desorbed. The second weight loss could be distributed to the decomposition of HPAV, which was completely decomposed at about 400°C. Compared with bulk HPAV, the temperature for the complete decomposition of the supported HPAV was 30°C lower. The result shows that dispersion of HPAV as a single molecule layer made its thermal stability decrease, which might be due to that there was not enough water or other counter ions in the secondary structure to support Keggin structure. Compared with HPAV/SBA and SBA-15, the weight loss of CsPAV/SBA was not that obvious. The first weight loss could be ascribed to the absorbed water (about 5%). Then the crystalline water lost slowly. It’s hard to figure out the crystalline water content on TG curves. CsPAV/SBA was decomposed completely at about 500°C which was about 100°C higher than HPAV/SBA, suggesting that the addition of Cs could significantly enhance the final decomposition temperature of the supported catalysts. With the increase of loading amount of CsPAV, the absorbed water content (about 3.5%) further decreased because of the decrease of surface area. 3CsPAV/SBA was completely decomposed at about 450°C. The results show that the addition of Cs in these supporting catalysts could obviously enhance thermal stability of the supported heteropoly catalysts.

Figure 6: TG-DTA curves of supported catalysts and SBA-15.
XPS: XPS was preformed to study the chemical changes of the catalysts during calcination and reaction. The chemical states of O, Mo, P, V and Si in the catalysts were detected and analyzed. O atoms in the catalysts play an important role in the selective oxidation of MAL to MAA [16]. The oxide species in the supported catalysts were analyzed by XPS, and two main kinds of oxide species were found (Figure 7). One was at 533 ± 0.25 eV which can be assigned to O species in SiO2, and the other was at 530 ± 0.2 eV assigned to O species in the supported CsPAV. The binding energy of these two O species in HPAV/SBA increased after calcination and decreased after catalytic reaction (Table S1). The increase of binding energy of O species was caused by the oxidation and decomposition during calcination, and the decrease of binding energy of O was due to the reduction by MAL during reaction. The decreased amount of O species in HPAV detected by XPS may be due to the decomposition of the supported HPAV. These two O species in CsPAV/ SBA showed the same tendency as HPAV/SBA due to the existence of HPAV in CsPAV/SBA. There were also two different O species in CsPAV (531.10~531.17 and 532.02~532.20 eV). These two O species showed different energy binding tendencies from the supported catalysts during calcination and reaction. The reduced one was the dominant species. The binding energy decreased after calcination and increased slightly after reaction, and the amount of reduced O species decreased after calcination and reaction. These changes were caused by the partial decomposition of catalysts during calcination and reaction, such as the migration of V atoms from the primary structure to secondary structure.

Figure 7: XPS spectra of O 1s, Mo 3d, V 3d, Si 2p in uncalcined, calcined, and used CsPAV/SBA.
XPS spectra showed that two different Mo species existed in the catalysts. The binding energy of first one was 233 ± 0.3 eV and that for the other was 232 ± 0.2 eV.
These two can be assigned to Mo species in Keggin structure in different chemical states. The chemical states of Mo in the catalysts were Mo5+ and Mo6+. Compared with the binding energy of HPAV/ SBA (233.30 eV), that of Mo in CsPAV/SBA (233.12 eV), CsPAV (233.17 eV) and 3CsPAV/SBA (233.20 eV) was lower (Table S2). It suggests that some supported HPAV was decomposed into MoO3 during preparation process, while the supported CsPAV was kept integral. The addition of Cs can prevent the decomposition of the catalysts during preparation process, and no Mo in low chemical state was found in CsPAV, calcined CsPAV and 3CsPAV/SBA. It suggests that the reduced chemical state of heteropoly compound was caused by the interaction between catalysts and supports. The amount of reduced Mo in HPAV/SBA and CsPAV/ SBA decreased after calcination due to the oxidation of Mo5+ by O2 during calcination. The amount of reduced Mo in all catalysts increased after reaction because of the oxidation of MAL on Mo-O species. No Mo species which belongs to MoO3 was detected by XPS and it may due to the reduction of Mo by MAL during reaction. Compared with CsPAV/SBA, less reduced Mo was found in 3CsPAV/SBA. It indicates that increasing loading amount can reduce the interaction between the catalysts and the supports.
Vanadyl species are also active species for this reaction [8]. XPS results showed that both V5+ (517.52~517.92 eV) and V4+ (516.65~516.98 eV) existed in the catalysts. The binding energy of V in the supported CsPAV and HPAV was also lower than that in bulk CsPAV, and more V5+ existed in bulk CsPAV than the supported CsPAV and HPAV. It also suggests that loading heteropoly compounds on SBA-15 can reduce their chemical states (Table S3). The amount of V4+ was decreased after calcination due to the oxidation by O2 in air. The amount of V5+ in the used HPAV/ SBA increased after reaction due to the decomposition of the catalysts during reaction. The decrease of V5+ in CsPAV/SBA after reaction suggested that the addition of Cs had improved thermal stability of the supported heteropoly catalysts and prevented the formation of V2O5.
Only one peak can be found at 103.8 eV in the Si spectra of SBA- 15 which can be assigned to the silica species in silica dioxide (Figure 7; Table S4), suggesting that only one kind of silica species was on the surface of SBA-15. After CsPAV being supported on SBA-15, the binding energy of silica shifted to 102.5 eV, and a new peak appeared at 100.2 eV. The changes of the binding energy showed that the supported CsPAV strongly interacted with silica species on the surface. The new emerged peak can be assigned to silica species surrounded by Mo atoms formed from the replacement of centre P atom by Si in Keggin structure. After calcination, the ratio of this kind of silica species decreased which suggested that some of the new formed Keggin structures with Si as centre atoms decomposed. The binding energy of the other silica species also increased to 103.5 eV which suggested that CsPAV interacted with the support and then decomposed during calcination. The binding energy of both of the two silica species decreased after catalytic reaction. The silica species emerged at 98.7 eV can be assigned to MoSix, indicating that the decomposed catalysts continued to react with the support during the catalytic reaction, and the rest of Keggin structures with Si as centre atoms were completely decomposed during the catalytic reaction. The XPS data suggests that the thermal stability of the catalysts decreased because of the interactions between the catalysts and the support which led the decomposition of the catalysts during calcination and reaction.
31P NMR and 29Si NMR: 29Si and 31P NMR spectra were collected to figure out the mechanism of decomposition of the supported CsPAV on SBA-15. Only one peak was shown in the 31P NMR spectra of CsPAV and the supported catalysts (Figure 8), suggesting that all the Keggin structures on the catalysts had the same state. However, the XRD patterns showed that some HPAV existed in the CsPAV/SBA. So there should be at least 2 peaks in the 31P NMR spectra of CsPAV/SBA. The only peak in 31P NMR spectra suggested that HPAV was well dispersed in the CsPAV. The chemical shift of 31P NMR spectra of bulk CsPAV (-5 ppm) was different from that of HPAV/SBA (-4.5 ppm) due to the replacement of protons by Cs+ in the catalysts [29-31]. After it was supported on SBA-15, the peak shifted upfield (CsPAV/SBA, -4.9 ppm; 3CsPAV/SBA, -4.8 ppm) was found, indicating that CsPAV could interact with SBA-15 during preparation. After calcination, the peak of P in CsPAV moved downfield (-5.2 ppm) because of the lost of crystalline water, but it shifted upfield for the other three supported catalysts, implying that the interactions between the catalysts and SBA-15 became stronger after calcination. The chemical shift of P in CsPAV was -5 ppm after the catalytic reaction due to the acquisition of crystalline water from the feed fluid. The peaks of P in the supported catalysts shifted downfiled after calcination and reaction, which indicated that Keggin structure interacting with SBA- 15 may decompose after the catalytic reaction. 29Si NMR was used to prove the results from XPS data and find out how the interactions occurred between the supported HPAV, CsPAV and SBA-15 (Figure 9). Two major peaks were found in the 29Si NMR spectra of the supported catalysts which can be assigned to (SiO)3=Si-OH (-102.5 ppm) and (SiO)3=Si-O-Si= (-110 ppm) [32,33]. These two peaks belonged to Si atoms in SBA-15. The new peak at -91 ppm can be found in the spectra of HPAV/SBA and calcined catalysts, which can be assigned to Si atoms in the Keggin structure. It indicated that SBA-15 interacted with HPAV during preparation and calcination, and the reaction between SBA- 15 and CsPAV only happened during calcination. The results were in agreement with other characteristic methods previously mentioned. The spectra suggested that some Si atoms entered Keggin structure of CsPAV and replaced P atoms. After reaction, disappearance of this peak suggested that the center atom of Keggin structures was replaced by Si.

Figure 8: 31P NMR spectra of uncalcined, calcined and used CsPAV and supported catalysts.

Figure 9: 28Si NMR spectra of uncalcined, calcined and used CsPAV and supported catalysts.
Reaction results
The effects of supports with different pore sizes: The above prepared catalysts were used to catalyze the oxidation of MAL to MAA. The catalytic activity of bulk CsPAV increased from 0.0016 to 0.019 min-1 with the reaction temperature increasing from 290 to 330°C and then decreased slightly (0.018 min-1) at 350°C (Figure 10). The activity of CsPAV/SBA increased from 0.013 to 0.026 min-1 with temperature increasing from 290 to 320°C and then decreased to 0.005 min-1 when temperature reached to 350°C. Catalytic activity of CsPAV/KIT increased from 0.008 to 0.017 min-1 (290-310°C) and then decreased to 0.004 min-1 (350°C).

Figure 10: The catalytic activity of CsPAV supported on different carriers in the oxidation of MAL to MAA (the unite of TOF is min-1, and the loading amount was 50%).
The catalytic activity of CsPAV/SiO2 decreased from 0.012 to 0.004 min-1 with reaction temperature increasing from 290 to 350°C, and the activity of CsPAV/Y decreased from 0.005 to 0.0015 min-1. The supported CsPAV showed higher catalytic activity than bulk CsPAV at 290°C. The selectivity of MAA on bulk CsPAV increased from 6.6% to 58.2% when the temperature increased from 290 to 330°C and declined to 47.1% at 350°C. For the supported catalysts, selectivity was much lower than bulk CsPAV (except at 290°C) and decreased with increase of reaction temperature (25% to 2.1% on CsPAV/SBA, 18.2% to 4% on CsPAV/KIT, 16.8% to 3.5% on CsPAV/SiO2 and 6.2% to 2.5% on CsPAV/Y). The highest MAA selectivity by using the supported catalysts was obtained on CsPAV/SBA, 25%. So SBA-15 was chosen for further study, and the catalytic activity was tested at 320°C. Bulk CsPAV was bright yellow before calcination and became chartreuse after calcination and turned to be dark green after catalytic reaction. The color changes of the catalysts may be due to the changes in chemical states of the elements. The green color might be caused by the reduction of Mo and V in the catalyst. For the supported catalysts, they had the same color as bulk CsPAV be fore and after calcination. They became brown after catalytic reaction, which may be caused by the decomposition of the supported CsPAV. So the decrease of catalytic activity and selectivity may be due to the decomposition of the catalysts. To enhance thermal stability of the catalysts, Cs content and loading amount were changed and the influence on catalytic activity investigated.
The supported catalysts with different Cs amount: The addition of Cs in the heteropoly catalysts could improve thermal stability and MAA selectivity [7,20]. Different amounts of Cs were added to the catalysts. The Brønsted acidity is necessary for the oxidation of MAL to MAA, so at least a proton should be left per heteropoly anion. The results showed that the addition of Cs decreased the catalytic activity (TOF), especially when the Cs content was higher than 1 per heteropoly anion (Tables 1 and 2). However, the selectivity of MAA was increased with the addition of Cs and it decreased significantly when the Cs content was higher than 1 per heteropoly anion. The results indicated that the preferential Cs content was 1 per heteropoly anion, and strong Brønsted acidity was needed for this reaction.
| Catalysts | BET surface area/m2·g-1 | Pore volume/cm3·g-1 | Average Pore width/nm |
|---|---|---|---|
| SBA-15 | 1017.8 | 1.307 | 6.6 |
| 0.5CsPAV/SBA | 569.4 | 0.822 | 6.4 |
| 1CsPAV/SBA | 450.5 | 0.734 | 7.01 |
| 2CsPAV/SBA | 236.7 | 0.349 | 6.9 |
| 3CsPAV/SBA | 108.5 | 0.121 | 6.6 |
| 4CsPAV/SBA | 86.2 | 0.187 | 7.33 |
| 5CsPAV/SBA | 70.0 | 0.149 | 7.4 |
| CsPAV | 5.8 | 0.021 | 16.40 |
Table 1: BET surface area, pore volume and pore width of the supported catalysts with differentsupporting amounts.
| Catalysts | TOF/min-1 | Selectivity% |
|---|---|---|
| HPAV/SBA | 0.028 | 20.4 |
| CsPAV/SBA | 0.026 | 25 |
| Cs2PAV/SBA | 0.007 | 7.1 |
| Cs3PAV/SBA | 0.002 | 4.5 |
Table 2: The catalytic activity and selectivity of supporting catalysts with different Cs contents at 320°C.
Catalysts with different loading amount: The activity increased from 0.026 to 0.042 min-1 with the loading amount of CsPAV increasing from 1 to 3 per gram of SBA-15, and then decreased to 0.023 min-1 on 5CsPAV/SBA, while the MAA selectivity showed a different trend, which increased from 25% to 44% with the increase of loading amount of CsPAV (Figure 11). Oxidation of MAL to MAA occurred on the active sites on the surface of catalysts. With the increase of loading amount of CsPAV, the sizes of CsPAV crystals became larger, which might show the similar properties with bulk CsPAV. So catalysts with high loading amount of CsPAV showed the similar selectivity as bulk CsPAV and the increase of loading amount could improve the selectivity. Compared with bulk CsPAV, the catalytic activity was improved more than twice on 3CsPAV/SBA. The suitable CsPAV loading amount of this catalyst was 80 wt%.

Figure 11: The catalytic activity and selectivity of 3CsPAV/SBA at 320°C with time on stream (TOF, bar; Selectivity, cubic point).
The catalytic life of the supported catalysts: Figure 12 showed that the catalytic activity kept steady in 12 hours, which increased slightly from 0.026 to 0.029 min-1 within the first 6 hours. Then it hold steady in next 6 hours. The selectivity increased from 27.6% to 37.1% with time going on, and then waved around 35%. No obvious deactivation was observed, which indicated that the addition of Cs could enhance thermal stability of catalysts.

Figure 12: The catalytic activity and selectivity of 3CsPAV/SBA at 320°C with time on stream (TOF, bar; Selectivity, cubic point).
According to the characterization results, changes of supported catalysts occurred during preparation, calcination and reaction. Firstly, Cs+ was well dispersed on the supports. After the Cs+ -containing supports were immersed into the HPAV solution, PMo11VO40 reacted with Cs and precipitated on the outer and inner surface of the supports. Due to the quick reaction of PMo11VO404- and Cs+, the reaction between PMo11VO404- process. If HPAV was supported directly on the supports, PMo11VO404- would react with SiOx species on the surface of the supports and led to decomposition of HPAV during drying.
Without the counter cations, HPAV in single molecule layer can easily interact with silica species and was decomposed by heating. Unlike HPAV dispersing on the supports in a single molecule layer, CsPAV clustered in small crystalline in the size of about 20 nm on the surface of the supports, and some catalysts molecules entered channels of the supports. With the support of other heteropoly anions, counter cation (mainly Cs+) and crystalline water, the catalysts showed better thermal stability than supported HPAV. However, there was some HPAV existed in the supported CsPAV, and it reacted with SiOx species on the surface during calcination and reaction. Si replaced P as the center atom in the heteropoly anions in Keggin structure, which caused the decrease of thermal stability and catalytic activity of the supported catalysts. The supported HPAV was decomposed into MoO3 and V2O5 which had good oxidization ability during calcination and reaction. So there were at least 3 kinds of oxidation catalysts on the supports, CsPAV, MoO3 and V2O5. According to the results of XRD, the crystalline sizes of MoO3 and V2O5 were rather small (smaller than 100 nm), so they would show very good catalytic activity. CsPAV is a good catalyst for oxidation of MAL to MAA, while MAL was oxidized to acetic acid and COx on MoO3 and V2O5. That’s why MAA selectivity on the supported catalysts was lower than bulk CsPAV.
With the increase of loading amount of CsPAV, the surface of SBA- 15 was covered by CsPAV, and MoO3 as well as V2O5 were not exposed directly to the feed. So MAL was oxidized on CsPAV, and the MAA selectivity was improved. The decomposed PMo11VO404- formed a layer of oxides between CsPAV and SBA-15 to avoid the further reaction between them during reaction and decomposition of supported CsPAV, so the catalytic activity and selectivity were steady with the reaction going on. In general, supporting heteropoly catalysts on SiO2 can reduce the size of the catalysts and improve the catalytic activity. The key point to prepare efficient supported heteropoly is to avoid the reaction between the catalysts and SiO2 supports and one way for that is to use a layer to separate catalysts and supports.
CsH3PMo11VO40 was supported on different kinds of SiO2 supports. The one supported on SBA-15 showed the best catalytic performance in the oxidation of MAL to MAA. CsH3PMo11VO40 formed clusters on the outer and inner surfaces of SBA-15. Compared with H4PMo11VO40 /SBA-15, the addition of Cs+ could improve thermal stability of the catalysts. The most suitable supporting amount of CsPAV was 80 wt%. The catalytic activity was improved twice than bulk CsH3PMo11VO40, but selectivity was low. The low selectivity was caused by MoO3 and V2O5 formed from the decomposition of the supported H4PMo11VO40 and CsH3PMo11VO40. The decomposition of CsH3PMo11VO40 and CsH3PMo11VO40 was due to the replacement of P by Si in Keggin structure. The decomposed catalysts formed an oxides layer between integral CsH3PMo11VO40 and supports which prevented any further reaction between CsH3PMo11VO40 and supports or decomposition of CsH3PMo11VO40. The key point to prepare efficient and thermal stable supported heteropoly is to avoid the reaction between the catalysts and silica supports.
The authors gratefully acknowledge the financial support of National Key Projects for Fundamental Research and Development of China (2016FB0600903) "Strategic Priority Research Program" of the Chinese Academy of Sciences (XDA07070600), National Key Projects for Fundamental Research and Development of China (2016YFB0601303), and National Science Funds for Distinguished Young Scholar (21425625).