Journal of Sleep Disorders & Therapy
Open Access
ISSN: 2167-0277
ISSN: 2167-0277
Review Article - (2013) Volume 2, Issue 5
Background: Persons who have sustained a traumatic brain injury are at a significantly increased risk for sleep disorders. One of the most commonly diagnosed sleep disorders after traumatic brain injury is sleep apnea, defined as a cessation of breathing accompanied by frequent arousals and hypoxia during sleep. The effects of untreated sleep apnea on a person’s cognitive decline and the development of behavioral deficits have only recently been identified. It has been shown that axonal damage can occur because of sleep apnea and numerous neuropsychological studies of sleep apnea patients show deficits in cognitive domains, such as executive function and attention. However, there has been little published discussion regarding the interaction between sleep apnea and executive function among persons with traumatic brain injury.
Objectives: The objectives of this review were to 1) review/synthesize published work relevant to the discussion of sleep apnea influencing executive function; and 2) clarify the nature of the interface between executive function and sleep apnea in persons with traumatic brain injury.
Results: Until now, little attention has been directed to the neurobehavioral consequences of sleep apnea in persons with traumatic brain injury. There is an urgent need for more longitudinal research examining the effects of sleep apnea on executive function after traumatic brain injury and the effectiveness of sleep apnea treatment on executive function after injury.
TBI: Traumatic Brain Injury; EF: Executive Function; SA: Sleep Apnea; CT: Computed Tomography; PET: Positron Emission Tomography; SDB: Sleep-Disordered Breathing; CSA: Central Sleep Apnea; OSA: Obstructive Sleep Apnea; RDI: Respiratory Disturbance Index; CNS: Central Nervous System; CPAP: Continuous Positive Airway Pressure; RCFT: Rey Complex Figure Test
Traumatic brain injury (TBI) is a major global health problem. According to the World Health Organization, TBI will surpass many diseases as the major cause of death and disability by the year 2020 [1]. Although the prevalence of TBI, defined as chronic symptoms from an earlier TBI, is not widely studied, it is estimated that in the USA, around 5.3 million people experience long-lasting symptoms caused by TBI [2]. If this statistic is extrapolated to a global scale, an oft-quoted estimate is that approximately 140 million persons are living with a TBI-related disability; however there is a possibility that this number is overestimated or underestimated [3]. Of the various disabilities that are the result of TBI, cognitive and psychological deficits, particularly poor executive function (EF), prevent TBI survivors from reintegrating into the community, returning to school, and re-entering the workplace [4]. Executive function, the cognitive ability that underlies planning, problem-solving, self-monitoring, behavioral control and working memory [5], is therefore a major consideration in the cognitive rehabilitation of patients with brain injury [5]. While a discussion of neurocognitive impairment secondary to TBI would seem to be straightforward, the pathogenesis is likely to be a multifactorial process involving a diverse range of mechanisms. Consequently, it is essential that an examination of post-morbid executive function be done with reference to the other common disorders that can affect the brain. In the current review, we provide an overview of published work that is relevant to the discussion of sleep apnea´s (SA) effects on EF and we examine different levels of evidence directed to clarifying the interface of executive function and SA in persons with TBI.
When the brain is injured, normal pathways for EF performance are disturbed. The organic disruptions as well as cerebral ischemia resulting from post-traumatic hypotension and hypoxia together contribute to the poor cognitive outcomes that often follow TBI [6]. Decreased cerebral blood flow after TBI has been revealed using different diagnostic tools, such as computer tomography (CT) and positron emission tomography (PET) scans [7-8]. The mechanisms by which post-traumatic cerebral ischemia occurs include injury to the vessels as a result of mechanical displacement, hypotension in the presence of auto regulatory failure, inadequate availability of cholinergic neuro transmitters, and potentiation of prostaglandininduced vasoconstriction. Because of an imbalance between cerebral oxygen consumption requirements and oxygen delivery, the brain tissue suffers from hypoxia [9]. It has been reported that the incidence, duration, and extent of tissue hypoxia is associated with poor neuropsychological outcomes post-TBI [10].
The phenomena of increased oxygen demand required for meta cognition and decreased oxygen delivery post-TBI are important concepts in understanding the underlying mechanism of EF decline after trauma to the brain. It is stipulated that persons with TBI who suffer from disorders causing decreased brain tissue oxygenation, constant or intermittent, due to other disorders (e.g.,SA), will demonstrate worse clinical outcomes post-TBI when compared to those who do not.
SA, an intermittent complete cessation of airflow during sleep, is the most frequently reported form of sleep-disordered breathing (SDB) after TBI. SA of at least 10 s is considered clinically important, but in many patients, apneas can last 20–30 s and in some cases as long as 3 min. SA can be central or obstructive in nature. In central sleep apnea (CSA), the neural drive to respiratory muscle is abolished. In obstructive sleep apnea (OSA), airflow ceases because of occlusion of the upper airway in sleep due to the specific anatomical structure of the upper airway or decreased muscle tonus resulting in muscle collapse during sleep. The resulting apnea leads to progression of asphyxia until there is an arousal from sleep, which restores the airway patency. The patient then returns to sleep and the sequence of the events repeats. In the most severe cases, SA occurs up to 500 times per night—resulting in sleep fragmentation and disturbances of sleep architecture. These nocturnal sleep disturbances result in vital exhaustion/daytime sleepiness/ impaired alertness and cognitive dysfunction, especially in the areas of attention and memory [11]. Furthermore, both OSA and CSA, highly prevalent after TBI, result in recurrent episodes of nocturnal asphyxia and decreased oxygenation (hypoxia) of brain tissue. Such brain tissue hypoxia is related to prefrontal lobe dysfunction [12]. Related respiratory events, hypopnea, and respiratory event-related arousals are characterized by a partial reduction in airflow in sleep, and consequent arousals from sleep with or without significant oxygen desaturation. The average number of all respiratory events per hour of sleep is termed the respiratory disturbance index (RDI). An RDI of 5 or greater is of clinical significance and can be an indication of SDB [13].
It is evident that SA may lead to a series of pathophysiologic events [14]. Xu et al. demonstrated that chronic intermittent hypoxia results in cortical neuronal cell apoptosis in mice [15]. Chronic intermittent hypoxia is also shown to be associated with inducible nitric oxide synthesis in the brain [16-17], which plays a role in nerve cell communication, and is associated with neuronal plasma membranes [18]. Other research has shown that free radicals formed as a result of oxidative stress in OSA lead to the up-regulation of transcription factors such as NF-κB [19]. Da Silva et al. reviewed the effects of OSA on homeostasis in the central nervous system (CNS) by examining the levels of biochemical markers of cerebral injury (neuron-specific enolase and the S100B protein derived from astrocytes) in OSA patients. The results of their study showed a significant increase in S100B markers, suggesting a CNS astrocyte reaction in response to cerebral hypoxemia in patients with OSA and supporting the view that OSA causes brain alterations, which can manifest as neuropsychological symptoms [20].
Numerous studies have established a relationship between SA and cognitive deficits, including EF, attention/vigilance, language, memory, and psychomotor speed [21]. Of these various deficits, EF has been a research topic of particular interest. Several tasks, such as the Wisconsin Card Sorting Task, the Tower of Toronto, and the Stroop task, have been used to measure SA sufferers’ deficits in EF [22].
Beebe and Gozal [12] studied the involvement of the frontal cortex in OSA patients and its link to EF. These authors reported decreased EF in their patients, which manifested as deficits in behavioral inhibition, self-regulation of affect and arousal, working and contextual memory, and analytical ability. They proposed a model in which sleep fragmentation, hypoxemia, and dysfunction of the frontal lobe cortex are interrelated through the disruption of homeostasis and altered glial viability within primarily frontal regions of the brain cortex. The authors stressed that executive dysfunction in OSA patients leads to reduced social relationships and job tasks; therefore, evaluation of social and work-related skills is crucial and should take precedence over other aspects of cognition (e.g., vocabulary, intelligence).
In one study, abnormal breathing and oxygen desaturation during sleep in heavy snorers were factors associated with obtaining fewer numbers of categories on the Wisconsin Card Sorting Task (i.e., domain of executive function: mental set shifting and abstract behavior). Using a modified version of the task, Naegele et al. [23] reported that poor performance on this task is predictive by the effect of severe hypoxemia in OSA patients. Behavioral manifestations in OSA patients also included difficulties with problem solving and initiation detected by the Tower of Toronto task (i.e., domain of executive function: problem solving). In addition, other researchers have found that persons with OSA require more time to complete EF tasks than do controls. When assessing planning abilities using the Maze task (i.e., domain of executive function: planning and foresight), Bedard et al. found that OSA participants had significant difficulties in completing the task [24].
Halbower et al. [25] studied links between OSA, cognitive deficits, and neuronal brain injury in children. Compared to controls, children with severe OSA had significant deficits in intelligence quotient (IQ) and EF (verbal working memory and verbal fluency), and demonstrated decreased neuronal metabolite ratios in the left hippocampus and the frontal cortex, as measured by proton magnetic resonance spectroscopic imaging. The authors speculated that untreated childhood OSA could permanently alter a developing child’s cognitive potential.
Several approaches to the treatment of SA have been advocated based on our current understanding of the mechanisms underlying the disorder. Milder OSA can often be managed effectively by weight reduction, avoidance of alcohol, improvement of nasal patency and avoidance of sleeping in a supine position. In more severe OSA cases, nasal continuous positive airway pressure (CPAP), a treatment which delivers positive pressure through a nasal/full-face mask to maintain opening of the upper airway during sleep, is widely used. Patients with CSA, whose apnea arises from instability of respiratory drive, have been shown to respond well to CPAP with or without adding nocturnal supplemental oxygen [26-27].
A current controversy regarding EF and treatment of SA revolves around an important issue: the efficacy of SA treatment in correcting executive dysfunction. Although newer research shows that cognitive deficits are resolved with treatment in the majority of cases, other research suggests some deficits in EF may remain unchanged [28-30]. This raises the possibility of permanent brain changes as a result of OSA.
In their review, Engleman et al. [31] indicated that most research to date has shown trends toward better cognitive performance after CPAP treatment compared to placebo. At least 3 studies comparing neuropsychological test scores of OSA patients before and after 6 months of CPAP treatment with test scores of healthy controls found notable cognitive improvement, including improvement in EF [23,32-33].
A recent randomized double-blind placebo-controlled trial by Ancoli-Israel et al. [34] examined the effect of CPAP on cognitive functioning in patients with Alzheimer’s disease (AD). A comparison of pre-and post-treatment neuropsychological test scores (HVLT-R, Trail Making test) in Alzheimer’s patients receiving 3 weeks of therapeutic CPAP showed a significant improvement in cognition, especially in episodic verbal learning and memory and in some aspects of EF such as cognitive flexibility and mental processing speed. These data, as well as the data of other researchers [35], seems promising and indicate that OSA may be a reversible cause of cognitive loss in this patient group; moreover, treatment of OSA, especially in the early stages of dementia when patients are still largely independent, may slow the progression of dementia. This reinforces the necessity of identifying and treating SA in individuals with TBI in the early stages post-injury.
Significant correlations have been reported between measures of EFs and regional cerebral glucose metabolism in medial and dorsolateral prefrontal cortical regions and the cingulate gyrus, despite the absence of detectable structural lesions on brain magnetic resonance imaging. These results suggest that impairments of EFs may be related not only to focal traumatic lesions of the prefrontal cortex, but also to lesions of the white matter tracts [36-37]. These effects appear to be non-specific to TBI, as similar results are reported in persons with Alzheimer’s disease AD [38]. Interestingly, SA is a risk factor for AD and other neurological diseases [39]. Similarly, the implication of TBI as a contributing factor in the development of AD and dementia has been reported [40]. In some individuals, disruption of axonal transport following TBI can lead to the rapid accumulation of amyloid precursor proteins and other proteins associated with the neurodegenerative disease. The variation observed in the APOE E4 allele in persons with TBI prompted exploration of the role of genetic factors in modulating the risk for AD after TBI [41]. Coincidentally, O’Hara et al. [42] reported the relationship between sleep apnea and dementia through the APOE E4 allele, whose carriers may be at an increased risk for developing dementia. Numerous research studies also report a higher prevalence of SA in the TBI population, ranging from 25% to 35%, compared to 4–9% in the general world population [43].
Although the high numbers of SA in the TBI population can be partly attributed to pre-morbid undetected sleep disorders, which can be a proximate cause of the injury itself, some persons may develop the sleep disorder as a consequence of brain injury. A disturbed coordination of upper airway and diaphragmatic muscles due to damage to the brainstem might favor the appearance of SA. Aggregation of SA can occur with commonly prescribed medications after the injury, such as muscle relaxants, sedatives, or hypnotics. Furthermore, narcotics for pain can cause opioid-induced respiratory depression, defined as a combination of decreased respiratory drive, reduced level of consciousness, and upper-airway obstruction, manifesting as both central and obstructive respiratory events [44].
Theoretical links exist between levels of hypocretins, which play a part in the regulation of various functions, including arousal, muscle tone, locomotion, feeding behavior, and neuroendocrine/autonomic functions, and SA [45-46]. Baumann et al. reported low hypocretin-1 levels in the cerebrospinal fluid from over 90% of 27 patients in the early days post-TBI and in 19% of 21 patients at 6 months post-injury [47-48]. Hypocretin deficiency after TBI may cause the loss of control of respiratory-related activity of hypoglossal motoneurons during rapid eye movement (REM) sleep [49]. Similarly, the presence of motor activity disfacilitation during REM sleep in acute rat preparations, with experimentally induced hypoxia, hypercapnia, and/or reduced airway patency has been reported; many of these conditions are also present in OSA [50]. Therefore, it is possible that due to TBI, disfacilitation of respiratory-related inputs to hypoglossal motoneurons during REM sleep will result in aggravation of SA at the acute stages post-injury, but may resolve with time.
Another relationship relevant to the discussion of executive dysfunction in SA and post-TBI is comorbid psychopathology. Depression after TBI is common, ranging 27–61%, when structural diagnostic criteria are used [51-52]. A nearly twofold increase in the risk for developing major depressive disorders has been reported in persons suffering from SA [53]. Working with acute mild TBI patients, Chaput et al. found that depression and irritability were more frequent in those with sleep complaints [54]. Depression has been reported to be associated with poorer cognitive outcomes, and no published studies have shown that the cognitive effects of depression can be reliably differentiated from the cognitive effects of SA or TBI [55]. Moreover, depression can act as an amplifier of other neurobehavioral changes, including aggression and irritability, and modulate symptom overlap between the post-concussion syndrome and psychopathology [56]. Given the frequent co-occurrence of depression and SA in TBI, recognizing the cumulative evidence for the intertwined processes of mood, sleep, and neuroplasticity post-TBI may have important implications for cognitive rehabilitation.
One of the aims of this review was to determine whether current evidence suggests that SA is causally associated with executive dysfunction after TBI and whether there is any evidence that the treatment of SA is effective in improving EF of these persons. The intent was to shed light on the potential importance of SA screening and treatment post-TBI as a means of halting the progression of executive dysfunction after brain trauma. To directly examine this question, prospective longitudinal studies are required. However, this research design is expensive and demands sustained commitment from participants and investigators. Given the heterogeneity in TBI (e.g., sex, age, injury localization and severity, confounding factors, difference in medication regime, and genetic makeup), the main requirement of an effective prospective study—the recruitment and retention of a sufficiently large sample—is difficult to achieve. An alternative approach has been to focus on comparisons of outcome between groups of persons with TBI of similar age/gender/time since injury/ injury localization/severity, but who differ in that some suffer SA and some do not.
Wilde et al. [57] performed such a study on 35 TBI patients, 18 years of age and older and at least 3 months post-TBI. Of these, 19 patients had OSA, as defined by polysomnography, with severity reported by the apnea–hypopnea index. Their EF performance was compared with 16 TBI participants without OSA. Both groups were comparable in terms of age, education, severity of injury, time postinjury, and Glasgow Coma Scale. The main outcome measures were: The Psychomotor Vigilance Test, Rey Complex Figure Test, Rey Auditory Verbal Learning Test, Digit Span Test from the Wechsler Memory Scale, and finger-taping test. The Rey-Osterrieth Complex Figure Test (RCFT) [58] permits the evaluation of different cognitive functions believed to be EF measures, such as visuospatial abilities, attention, planning, organizational skills and analysis-synthesis. Persons with OSA performed significantly worse on RCFT delayed recall (t35= -2.05, p=0.048), had greater numbers of lapses, and worse retention (i.e., deterioration of thinking ability and memory) compared to those without OSA. Interestingly, the researchers did not find significant differences in OSA vs. non-OSA patients on working memory tests such as the copy administration, total recall list, and the digit span test forward and backward (Table 1). These results are promising, suggesting the possibility of reversibility of executive deficits and the potential of recovery with treatment implementation.
| Author(s); year | Article Title, journal, pages; Study Objective | Research Design/ Sample Size; Inclusion/Exclusion | Participants | Data Collection Methods/Measures | Findings | Comments |
| Wilde M, Castriotta R, Lai J, Atanasov S, Masel B, Kuna S; 2007 [57] |
Cognitive impairment in patients with traumatic brain injury and obstructive sleep apnea. Arch Phys Med Rehabil. 2007; 88(10):1284-8 To examine the impact of comorbid obstructive sleep apnea on the cognitive functioning of traumatic brain injury patients |
Case-control study/35; subjects who were more than 18 years old and at least 3 months post injury/ Subjects with other sleep disorders | Persons with traumatic brain injury (TBI) from 3 academic medical centers; 2 groups: obstructive sleep apnea (OSA) and non-OSA; n (OSA) =19, n (non-OSA) = 16 Age: mean for OSA, 47.25 y; non-OSA, 51.47 y Time since injury: OSA, 124 mo; non-OSA, 77 mo Male/Female: OSA, 17/2; non-OSA, 12/4 TBI severity (unknown/moderate/moderate-severe/severe): OSA, 10/3/1/5; non-OSA, 8/2/3/3 Computed tomography (CT) findings (unknown/negative/positive): OSA, 16/0/9; non-OSA, 8/1/7 |
Assessments: Clinical interview; Physical examination; Glasgow Coma Score (GSC); CT data; Neurological assessment; Polysomnography (PSG); Multiple Sleep Latency Test (MSLT) |
Mean apnea–hypopnea index (AHI) for OSA group: 26.83/h; non-OSA: 4.49/h Mean MSLT score: OSA, 11.19; non-OSA, 5.06 Statistically significant difference in test performance for patients with and without OSA: • memory recall measures: worse in OSA • number of lapses: greater in OSA • retention: worse in OSA No statistically significant difference in performance between groups: • on the copy administration • total recall list • digit span test forward and backward |
Definitions: Diagnosis of OSA: 5 or more apneas per hour and/or 10 or more apneas plus hypopneas per hour |
Table 1: Literature review on association between traumatic brain injury and sleep apnea.
The study has noted limitations. Some data were missing in 10 of 19 participants with OSA and in 8 of 16 non-OSA participants. The localization of brain damage was not reported, even though positive CT scan findings were found in 9 of 19 OSA and 8 non-OSA participants. The authors did not comment on the number of CSA events or whether any hypopneas that were included in the diagnoses were of central origin. Overall, this is valuable information because SA in TBI populations seems to differ from those being described in general. According to reports of Websters et al. [10], the majority of the events in their sample of 28 TBI participants were central in nature rather than obstructive, while in the general population, SA consists of 90% obstructive and only 10% central events [59]. Additional methodological concerns include the great number of variables and the small sample size consisted largely of male participants, which was not allowing definitive conclusions to be drawn.
In summary, executive dysfunction, encompassing impairments in cognitive, behavioral and emotional processes after TBI, presents significant challenges for recovery after injury. Similar process impairments are also found in persons with SA. This review suggests that undetected/untreated SA in persons with TBI may have profound effects on neurobehavioral processes, impacting both structure and function. Different lines of evidence are relevant to the mechanism of EF decline associated with TBI and untreated SA. These include, but are not limited to: 1) evidence from animal studies on hippocampal cell death and apoptosis of the forebrain regions as a result of recurrent SA resulting in secondary neuronal injury, 2) association between white-matter grade and severity of CSA reported, 3) results of CPAP treatment intervention studies showing improvement in EF test performance in SA sufferers, 4) studies on the prevalence of SA in TBI populations, placing affected individuals in a category of greater risk, 5) numerous studies showing the detrimental effects of SA on non-TBI persons’ cognitive performance, especially EF, all of which advocate for an association between possible undiscovered and untreated SA and poor cognitive rehabilitation outcomes of TBI survivors. Finally, a recent study by researchers from the University of Pittsburgh School of Medicine retrospectively evaluated diffusion-tensor images from 64 consecutive patients with mild TBI who underwent magnetic resonance imaging. They found that more than half of the patients had sleep and wake disturbances. While researchers did not clarify the category of the sleep disorder, there was a significant reduction in fractional anisotropy in the parahippocampalgyri in patients with sleep and wake disturbances versus those without [36]. This region in the brain plays an important role in memory encoding and retrieval and in identifying social context as well, including paralinguistic elements of verbal communication [60].
Although we have presented a narrative review of published literature relevant to understanding the association between SA and executive function in TBI, a significant challenge in discussing these associations is disentangling the effect of brain injury from the effect of SA on executive function in TBI (Figure 1). It is possible that studies of executive function have been conducted often with TBI survivors who have suffered SA, where untreated pre-existing SA was a proximate cause of the brain injury itself. There is in fact growing evidence supporting an association between sleepiness and fatigue as a result of SA, and the probability of one’s involvement in an incident involving injury [60]. As discussed above, non-specific alterations to white and grey matter integrity and aberrant prefrontal activity have been reported in both TBI and SA sufferers’ studies. These provocative findings suggest a need for further study of brain connectivity among areas particularly relevant to executive function and vascular risk factors, taking into account the high likelihood of undetected/untreated SA in TBI persons. Given that there is no known cure for brain injury, addressing SA may provide benefits in terms of cognitive function which may be currently overlooked.
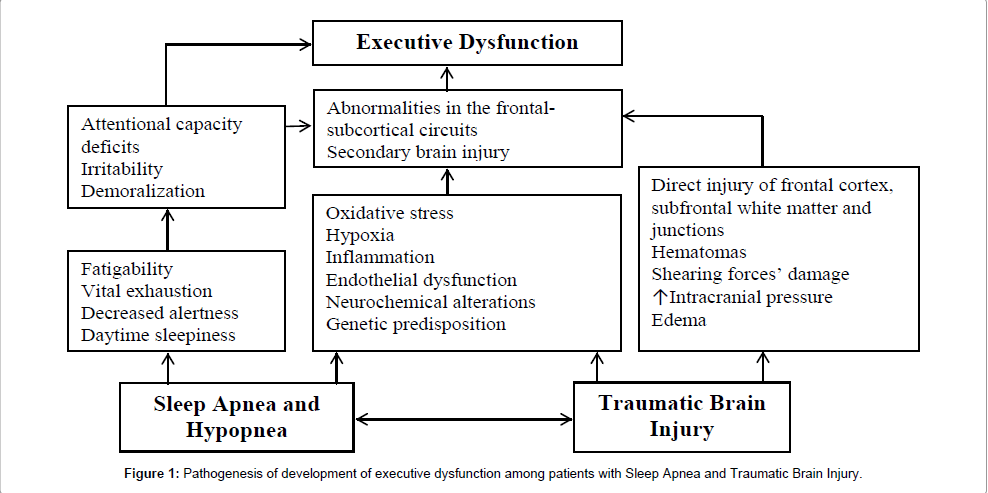
Figure 1: Pathogenesis of development of executive dysfunction among patients with Sleep Apnea and Traumatic Brain Injury.
To conclude, the findings of this review suggest that limited research that has been done to identify EF outcomes in persons with TBI, and how these outcomes might be related to SA. Considering the impact of brain damage itself on cognitive functioning after TBI, it is still unknown whether executive dysfunction related to SA in the TBI population is a specific entity or whether the presence of SA exaggerates the TBI-related physiological changes in EF. The general processes slowing usually found in patients with brain injuries could explain, to a great extent, the decline in EF observed in persons with TBI, as well as persons with SA [14, 37-40]. More definitive evidence on SA as a cause of poor EF after TBI can be gathered through further research targeting TBI survivors. While it is premature to make strong conclusions regarding the nature of the synergy of SA and executive dysfunction in TBI, this literature review sheds light on the repercussions of SA, if left unrecognized and untreated, to cognitive dysfunction following brain damage. The prospect of improving impaired processes through the treatment of SA cannot be ignored. The current state of evidence indicating a hierarchy of post-morbid cognitive symptom clusters within similar injury severities, as well as sustained executive dysfunction after mild TBI years after injury, further strengthens the need to identify and treat people with TBI who experience SA at the early stages of cognitive rehabilitation.
This manuscript had no external funding source. The first author was supported by a 2012/13 Toronto Rehabilitation Institute Scholarship, 2012/13 Ontario Graduate Scholarship, and 2013/15 Frederick Banting and Charles Best Canada Graduate Scholarships-Doctoral Awards from the Canadian Institutes of Health Research. We also recognize the support of the Canadian Institute of Health Research - Institute of Gender and Health #CGW-126580.