Pancreatic Disorders & Therapy
Open Access
ISSN: 2165-7092
ISSN: 2165-7092
Review Article - (2014) Volume 4, Issue 3
Solid Pseudopapillary Neoplasms (SPN) of the pancreas is rare tumors found throughout the pancreas composed of solid and cystic components. Since their description by Frantz, SPNs have been shown to comprise 1-2% of pancreatic tumors. These neoplasms predominantly affect young females in the second and third decades of life. Little is known about the pathogenesis of these tumors, although there is suggestion of a neuroendocrine origin or relationship with sex hormone receptors. The mainstay of treatment continues to be surgical resection, even in patients who present with locally advanced or metastatic disease. SPNs have low malignant potential with 5-year survival estimated as high as 95%, including those with malignant disease. We aim to review the current understanding regarding the diagnosis, management, and outcomes of patients with solid pseudopapillary neoplasms of the pancreas.
<Keywords: Solid pseudopapillary neoplasms, Chemoradiation, Pseudopapillae, Alpha-1-antitrypsin
Solid Pseudopapillary Neoplasms (SPN) of the pancreas is rare tumors of low malignant potential composed of solid and cystic pseudopapillae which predominantly affect young females [1]. The first documentation of this lesion was by Lichtenstein in 1933, who described a young female presenting with a large abdominal tumor from which she ultimately died. At necropsy, she was found to have a large encapsulated cystic tumor of the pancreatic tail in addition to metastases to the liver, omentum, and peritoneum [2-5]. The patient was diagnosed as having a papillary cystadenocarcinoma; however, recent pathological review equates the findings most consistent with a mucinous cystic neoplasm. The first indisputable description of an SPN is attributed to Frantz, who in 1959 described a papillary tumor of the pancreas with solid and cystic components [6]. After this report, SPNs were described in several other case reports under a variety of names descriptive of its assorted characteristics, including “papillary epithelial neoplasia,” “Frantz tumor,” “papillary-cystic neoplasm,” and “adenocarcinoma of the pancreas in childhood” [7-9]. It was not until 1996 that the World Health Organization specifically classified the disease as “solid pseudopapillary tumor of the pancreas,” combining its different characteristics under one diagnosis [10].
In general, SPNs are solitary, round, well circumscribed tumors composed of intermingled solid and cystic areas, often with a hemorrhagic component. Tumor size differs widely amongst patients. Mean diameter of tumor was shown to be 8.6 cm in a series of over 2700 patients, while two smaller series placed the mean diameter at 6.08 cm (range 0.5 to 34.5 cm) and 7.87 cm (range 1-25 cm) [1-3]. Tumor appearance will vary based on size, as smaller tumors are often less cystic, unencapsulated, and may be less well circumscribed compared to larger tumors [11,12]. By histology, SPNs demonstrate alternating components of solid regions with sheets of uniform epitheliod cells and cystic spaces with pseudopapillae and necrosis, often surrounded by a fibrous capsule [12,13]. A pseudopapillary pattern is seen as tumor cells outgrow their blood supply and break apart, leaving cystic spaces containing hemorrhage, foamy macrophages, and cholesterol clefts as degenerative changes occur [12,14]. The cells often have abundant cytoplasm, with round to oval nuclei with frequent indentations, groves, and membrane irregularities. Mitotic figures and pleomorphism are generally absent from the nuclei, although some atypia may be seen due to degenerative changes [12,13]. Despite the typical presence of a fibrous capsule, it is not uncommon to see extension of the tumor through the capsule into the surrounding normal pancreatic tissue [12,14].
Despite a diverse immunophenotype, immunohistochemistry can be useful in differentiating SPNs from other pancreatic tumors. One of the most useful markers for SPNs is β-catenin. Studies have shown that SPNs often have mutations in the β-catenin pathway, leading to excess accumulation of the β-catenin protein in the nucleus not seen in other pancreatic neoplasms [1,15,16]. Other markers that consistently stain in the majority of SPNs include CD10, vimentin, Alpha-1-Antitrypsin (AAT), progesterone receptors, and Neuron-Specific Enolase (NSE) [17-19]. However, the majority of Pancreatic Neuroendocrine Tumors (PNET) also stains for vimentin, NSE and AAT, making them relatively non-specific for SPNs [12,18]. Unlike PNETs, SPNs are usually nonreactive for chromogranin with a small percentage of patients staining weakly-positive to positive for synaptophysin [12,17,18]. Finally, SPNs have been shown to stain for cytokeratin, although positivity is often weak and ranges dramatically from 10% to 74% based on the study making it an unreliable diagnostic marker [12,17,20].
The pathogenesis of SPN is currently unknown. Immunohistochemical staining of SPNs with predominantly neuroendocrine markers such as vimentin, NSE and AAT have suggested a neuroendocrine origin [17-19,21]. However, staining is not seen in SPNs for more specific neuroendocrine markers such as chromogranin A, making an argument against neuroendocrine differentiation [12]. Given the predominance of SPN in young females, some have suggested a relationship with gender hormone receptors. With rare exceptions, no study has demonstrated the presence of estrogen receptors in these tumors [12,17,22-24]. While a study of two patients demonstrated binding of SPN cellular lysates to 3H-estradiol suggesting the presence of estrogen receptors, this binding was also seen with normal pancreas [25]. The presence of progesterone receptors in most if not all SPNs have led to the theory that the reproductive period may stimulate growth of these tumors, leading to their predilection in young females. However, staining for the progesterone receptor has been shown in both males and females, casting some doubt on this hypothesis [12,17,19,24,26]. In addition, no study has shown a dependence of SPN on estrogen or progesterone receptors for growth.
Solid pseudopapillary neoplasms are overwhelmingly a diagnosis of young females. The largest review to date of 2,744 patients with SPN found almost 88% were female [3] (Table 1). Smaller reviews of 718 patients and 533 patients found 91% and 89%, respectively, of SPNs diagnosed in females, leading to a male to female ratio of between 1:8.4 and 1:9.7 [1,2]. In general, SPNs are predominantly diagnosed during the second and third decades of life. Median age at diagnosis ranged between 21.97 years to 28.5 years [1,3]. The majority of patients are symptomatic at the time of diagnosis, and the most common symptom noted is abdominal pain [1-4]. Similar to other pancreatic tumors, most symptoms are non-specific and include nausea or vomiting, abdominal discomfort, or discovery of a palpable abdominal mass [1,3]. Jaundice and pancreatitis tend to be relatively rare symptoms. Approximately 20% to 30% of patients are asymptomatic and are diagnosed incidentally during a work-up and imaging for other reasons [1,3]. There does not appear to be any association with specific racial or ethnic groups, and no link has been identified to an inheritable genetic syndrome [2,4].
| Papavramidis1 | Yu2 | Law3 | |
| Patient number | 718 | 553 | 2744 |
| Female sex | 0.907 | 0.893 | 0.878 |
| Male:Female Ratio | 0.000807639 | 0.000791319 | NR |
| Mean Age (yr) (Range) | 21.97 (2 – 85 years) | 27.2 (6 – 71 years) | 28.5 (SD, 13.7 years) |
| Mean size (cm) (Range) | 6.08 (0.5 – 34.5 cm) | 7.97 (1 – 25 cm) | 8.6 (SD, 4.3 cm) |
| %Local or Vascular Invasion | NR | NR | 0.046 |
| %Metastases | 19.5% (includes local invasion) | 9.2% (includes local invasion) | 0.077 |
| %Recurrence | 0.066 | 3.6% (local only) | 0.044 |
| 5-year Survival | 0.95 | 0.969 | NR |
Table 1: Demographics for patients with SPN from the three largest systematic reviews. Key: NR=Not reported; SD=Standard Deviation.
SPNs can be found throughout the pancreas but in general appear to have a predilection for the head and tail (Figures 1 and 2). The largest review to date which included 2,744 patients showed 1,626 (59%) localized in the body and/or tail and 988 (36%) in the head of the pancreas [3]. An analysis of 718 patients showed almost equal prevalence of SPN in the tail (26%) and head (34%) of the pancreas, with the remaining tumors in the body (15%), body and tail (10%), and neck (1%) [1]. A smaller review of 553 patients showed the most common tumor sites to be the pancreatic head (49.8%), tail (24%), body and tail (19.5%) and body (11%) [2]. Approximately 1-2% of SPNs are found outside of the pancreas, with common sites including the adrenal, retroperitoneum, and mesentery [1-3].
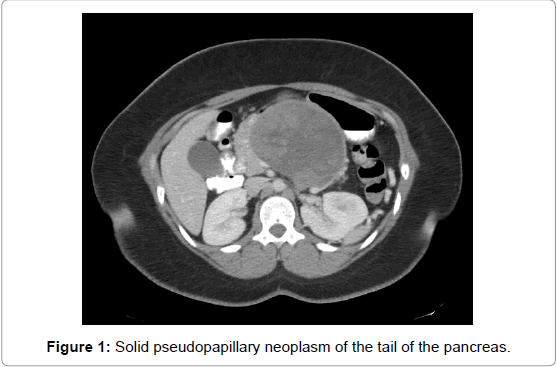
Figure 1: Solid pseudopapillary neoplasm of the tail of the pancreas.
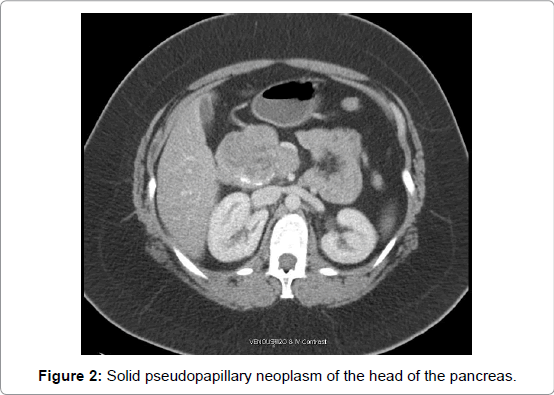
Figure 2: Solid pseudopapillary neoplasm of the head of the pancreas.
Current data suggests a low rate of metastases and vascular invasion in SPN. In general, it is estimated between 5-20% will present with local invasion or metastases [1,13]. Of the individual case studies providing detailed pathologic staging on a total of 1,523 patients, vascular involvement was shown in approximately 5% of patients, with lymph node and distant metastases in 1.6% and 7.7%, respectively [3]. A smaller analysis of 497 out of 718 patients with data on metastases, 19.5% (n=97) were found to have local invasion or metastases [1]. In these patients, the most common sites were the liver and portal vein (27 and 26 patients, respectively), followed by the spleen (17 patients) and smaller numbers to other organs (omentum, blood vessels, duodenum, colon, and lung) [1]. Similar metastatic locations were shown in the 45 (9.2%) of 491 patients who presented with malignant SPN, the most common being invasion into the portal, splenic or superior mesenteric vein followed by duodenum, peritoneum, liver, and spleen [2]. Lymph nodes were found to be positive in 3 of these patients [2]. Current data does not suggest any factors associated with the diagnosis of metastatic SPN.
The most common and useful imaging for diagnosis of an SPN is Computed Tomography (CT) [1,3]. CT scans will usually show a large, encapsulated heterogeneous mass of the pancreas with solid, cystic, and hemorrhagic components in addition to rare calcifications, although they will less commonly present as mostly cystic or mostly solid [27]. CT scan also allows for the assessment of vascular invasion and distant metastases before intervention are undertaken. The most common type of diagnostic workup was CT scan, accounting for 49% and 83% of imaging in two large studies [2,3]. The second most common form of imaging was transabdominal ultrasound, followed by Magnetic Resonance Imaging (MRI) in 7.8%-12 % of patients [2,3]. The largest review of 2,744 found documentation of 253 patients who underwent preoperative biopsy, either percutaneously (58.5%) or EUS-guided fine needle aspiration (41.5%); SPN was correctly identified by biopsy in approximately 65% of cases [3]. Conversely, of the 325 patients who underwent preoperative biopsy in another review, only 77 (23.7%) were correctly diagnosed or suspected of having an SPN [2].
The first surgical resection of an SPN was performed in 1970 by Grosfeld as described by Hamoudi, and since that time surgical resection has been the mainstay of treatment [7]. The specific operation depends on the location of the SPN, although the appropriate option for resection has not yet been determined. The majority of patients underwent either a distal pancreatectomy (40% to 48.6%) or pancreaticoduodenectomy (Whipple operation, 21.5% to 25%), with a smaller portion undergoing a central or total pancreatectomy [1,3]. A significant number of patients in all three studies underwent enucleation of their tumor. While this number was low in two of the studies (4.7% and 8.8%, respectively), 104 patients, or 33%, in the Chinese series underwent local resection or enucleation, making it the most common resection [1-3]. As with other pancreatic tumors, concern remains that microscopic tumor cells could be left behind, leaving the patient at higher risk for recurrence. At this time, it is uncertain how this relates to overall survival and recurrence of disease, although limited reports suggest that patients with a microscopic or grossly positive margin following resection did not develop a recurrence or have decreased survival [4,28-30].
With reports of as many as 20% of patients presenting with metastatic disease, local invasion, limited metastases, or disease recurrence are not felt to be contraindications to surgery. However, it remains controversial given the concern of recurrence given mixed long-term results. In one small series, four patients who presented with metastatic disease underwent resection of their primary lesion and liver metastases [29]. Of the four patients, one died at 8 months due to progressive metastatic disease. Of the remaining three patients, one was alive with recurrent disease at 6 years while the other two were alive without evidence of recurrence at 6 months and 11 years. Other small series of patients have shown some long-term disease free survivors with others developing recurrences [31-33]. Given studies have shown prolonged survival after resection compared to those patients who do not undergo surgery, resection in appropriately selected patients with metastatic disease at selected centers is feasible.
Data is limited on the role of chemotherapy and radiation for the treatment of SPN, although studies suggest it is successful in selected patients. Large studies have shown only a small minority of patients will undergo adjuvant therapy. Of the studies that reported on chemoradiation therapy, only 6.3% of patients were reported to have undergone adjuvant therapy; this was split between chemotherapy (4.7%) and radiation therapy (1.6%) [3]. The most commonly used chemotherapeutic agents were 5-Fluorouracil (5-FU) and Gemcitabine. Given the benefits of surgical resection, the primary utility of chemoradiation is in patients who present with unresectable disease. A few studies involving a small number of patients have shown the use of preoperative chemotherapy or radiation therapy can shrink the tumor and allow for resection or prolong survival [34-36]. However, this data remains anecdotal and further study is needed.
Outcomes are generally excellent for patients after resection of an SPN, especially compared to survival for other types of pancreatic neoplasms. The majority of patients who undergo surgical resection are cured of their disease. The largest review of patients with SPN reported almost 96% were disease free after a follow-up period of 36.1 months [3]. Of the 4% of patients in this study who had disease recurrence, the median time to recurrence was 50.5 months [3]. Two other large reviews showed disease recurrence of 3.8% and 6.6%, most commonly to the liver and lymph nodes [1,2]. Other studies have shown that patients with “unresectable” disease can survive for over ten years without surgery. Overall, patients with an SPN have estimated 2 and 5 year survival rates of 97% and 95%, respectively [1]. Of the 3.1% of patients who died in the study, only half of the deaths were attributed to SPN.
Solid Pseudopapillary Neoplasms (SPN) of the pancreas is solitary, well circumscribed tumors composed of intermingled solid and cystic areas, often with a hemorrhagic component [1]. These tumors are rare and comprise only 1-2% of all pancreatic tumors, but predominantly affect young females in the second and third decades of life [1-3]. Little is known about the pathogenesis of these tumors, although there is suggestion of a neuroendocrine origin based on staining for markers such as vimentin, NSE, and AAT [17,18]. In addition, there is a suggested correlation with sex hormone receptors based on the female predominance, although data does not support this theory. The mainstay of treatment continues to be surgical resection, even in patients who present with locally advanced or metastatic disease [1,3]. Chemotherapy and radiation are often limited to those with unresectable disease in order to shrink tumors and allow for surgery. The majority of patients are cured after resection, with recurrence noted in only 3.8% to 6.6% of patients [1-3]. SPNs have low malignant potential with 5-year survival estimated as high as 95%, including those with malignant disease [1,4]. Despite the overall good prognosis of this disease, further research is needed to better predict aggressiveness of disease and survival while better defining the role of systemic therapy.