Journal of Theoretical & Computational Science
Open Access
ISSN: 2376-130X
ISSN: 2376-130X
Research Article - (2015) Volume 2, Issue 4
In this attempt of research work, the inducement of NLO property on the compound Acetone thiosemicarbazone has been analyzed using computational calculations. The FT-IR, FT-Raman, FT-NMR and UV-Visible spectra have been recorded in specified region. The optimized inducement of NLO activity by the molecular structural deformation due to the addition of acetone compound has been investigated. The supportive analyses such as Mulliken charge levels, first order and second order polarization, vibrational confirmation, frontier molecular interactions, thermodynamic function (Gibbs energy) and VCD profile for proving NLO mechanism in the compound have been carried out. The chemical environment of the compound was simultaneously monitored by simulating and recording 1H and 13C NMR spectra. The isotropic and anisotropic chemical shift related to carbons and hydrogens after the formation of target compound have been carefully interpreted. The stabilization of orbitals by interchanging of energy between donor and acceptor was observed by NBO perturbation calculations.
<Keywords: Acetone thiosemicarbazone, NLO activity, VCD, Isotropic, NBO perturbation
Molecular materials especially, organic materials with nonlinear optical (NLO) properties are presently attracting considerable interest because of their potential applications in the optoelectronic devices of data communications, information storage, optical computing, signal processing [1,2] and terahertz (THz) wave generation [3]. At present, a wide range of stabilized HOMO (Donor) and LUMO (acceptor) substituted organic compounds are being investigated to emphasize the relationship between molecular structure and non linear response. Especially organic structures with large delocalized π-systems are more easily affected by an external optical field as they are relatively loosely bound to the nucleus, and that the delocalized orbitals may be extended over the entire molecule giving large and fast polarization [4,5]. These delocalized π-systems results spatial asymmetry of the electron distribution which is directly associated with Second order NLO-effects [6]. In nonlinear optics, first and second order hyperpolarizabilities are the heart of consideration because of their importance for the production of new materials and devices in opto-electronic industry [7,8].
The thiosemicarbazone is a favored class of compounds of optical chemistry research for recent years because of their crystal structure lacks a centre of symmetry, stability, their optical activity, and their reactivity [9-12]. The thiosemicarbazone and its derivatives are very interesting and have gained special attention in non liner optical application due to their unusual non centro symmetry. As the base compound with potential sulfur and nitrogen donors and hydrogen acceptors, the change in dipole moment and the energy difference between them are causing the second order hyperpolarizability in the compound. Further substitution of CH3 and other groups in the thiosemicarbazone chain generates functional delocalized π-electron systems such as imine (C=N), thio (C=S) and azine groups (NN), among which the imine represent a most prominent group. It is interesting to see how the properties change upon this specific subtution of couple of CH3 fragments as acetone.
After screening the literatures, it was found that, no vibrational and NLO analytical work has been made in the complex system of acetone thiosemicarbazone. In this work, the crystal was fabricated and the important molecular spectra were recorded to evaluate the inducement of optical and NLO properties in the compound. The suitable computational calculations of HF and DFT have been performed and the results obtained in the experimental techniques were evaluated.
Preparation of acetone thiosemicarbazone
The organic crystal of acetone thiosemicarbazone was prepared by adopting general procedure [13,14]. In a hot solution of thiosemicarbazide, an appropriate amount of methanol was added by drop wise and in the solution of the acetone, measured quantity of methanol during thirty minutes. The mixture was thoroughly stirred and refluxed for four hours. Then, concentrates was filtered half the volume under reduced pressure. The concentrate was allowed for slow evaporation at room temperature. The crystals were collected by filtration and washed with cold ethanol then dried. The grown crystals were purified by repeated- recrystallization.
Spectroscopic measurements of FT-IR, FT-Raman, NMR and UV-Visible spectra
• FT-IR, FT-Raman spectra were recorded in Bruker RFS 27 spectrometer with full specification.
• UV-Visible spectrum was recorded using Lambda 25 Spectrometer with latest specification.
• The 1H and 13C NMR spectral data was obtained from BRUKER 300 FT-NMR spectrometer.
The calculations of optimization for structural parameters and atomic charges of the title compounds were carried out using the Gaussian 09 D .01 program package [15,16] by using DFT and HF with 6-311++G(d,p) basis set. The optimized parameters were viewed and evaluated from the Gauss view 09 D .01 version [17] and were given in the Table 1. The electronic absorption spectra obtained among the frontier molecular orbitals provides an explanation and estimation of the Kubo gap of the title molecular system and were calculated at B3LYP/6-311++G(d,p) level which exposes the details of chemical reaction path within the molecule. The restricted basis sets are completely described the wave functions in the most popular quantum method in terms of electron density and other properties that were depicted by the possibility of filled NBO. The interaction between bonding and anti-bonding (Rydberg orbitals) signified the divergence of molecular structure from the Lewis model and it was used to measure the delocalization [18,19].
| Bond Parameters | HF/6-311++G(d,p) | B3LYP/6-311++G(d,p) | Experimental |
|---|---|---|---|
| Bond Lengths (Å) | |||
| C1-C2 | 1.502 | 1.501 | - |
| C1-C6 | 1.511 | 1.509 | - |
| C1-N10 | 1.257 | 1.284 | 1.287 |
| C2-H3 | 1.087 | 1.095 | - |
| C2-H4 | 1.081 | 1.089 | - |
| C2-H5 | 1.087 | 1.095 | - |
| C6-H7 | 1.081 | 1.089 | - |
| C6-H8 | 1.086 | 1.096 | - |
| C6-H9 | 1.086 | 1.096 | - |
| N10-N11 | 1.361 | 1.364 | 1.384 |
| N11-H12 | 0.994 | 1.012 | - |
| N11-C13 | 1.344 | 1.371 | 1.346 |
| C13-S14 | 1.683 | 1.677 | 1.669 |
| C13-N15 | 1.328 | 1.346 | 1.326 |
| N15-H16 | 0.991 | 1.004 | - |
| N15-H17 | 0.993 | 1.009 | - |
| Bond Angles (°) | |||
| C2-C1-C6 | 117.814 | 118.507 | - |
| C2-C1-N10 | 117.654 | 117.474 | - |
| C6-C1-N10 | 124.531 | 124.018 | - |
| C1-C2-H3 | 110.146 | 110.614 | - |
| C1-C2-H4 | 110.662 | 110.70 | - |
| C1-C2-H5 | 110.146 | 110.614 | - |
| H3-C2-H4 | 109.230 | 108.961 | - |
| H3-C2-H5 | 107.355 | 106.890 | - |
| H4-C2-H5 | 109.230 | 108.961 | - |
| C1-C6-H7 | 110.653 | 111.012 | - |
| C1-C6-H8 | 110.979 | 111.336 | - |
| C1-C6-H9 | 110.979 | 111.336 | - |
| H7-C6-H8 | 107.995 | 107.743 | - |
| H7-C6-H9 | 107.995 | 107.743 | - |
| H8-C6-H9 | 108.115 | 107.488 | - |
| C1-N10-N11 | 118.889 | 118.671 | 115.1 |
| N10-N11-H12 | 122.624 | 123.066 | - |
| N10-N11-C13 | 121.132 | 121.227 | 119.3 |
| H12-N11-C13 | 116.243 | 115.706 | - |
| N11-C13-S14 | 120.133 | 120.265 | 119.5 |
| N11-C13-N15 | 116.540 | 115.185 | 117.5 |
| S14-C13-N15 | 123.326 | 124.548 | 122.9 |
| C13-N15-H16 | 118.077 | 118.322 | - |
| C13-N15-H17 | 120.799 | 120.071 | - |
| H16-N15-H17 | 121.122 | 121.605 | - |
| Dihedral Angles (°) | |||
| C6-C1-C2-H3 | 59.1162 | 59.1196 | - |
| C6-C1-C2-H4 | 180.0002 | 180.0001 | - |
| C6-C1-C2-H5 | -59.1157 | -59.1194 | - |
| N10-C1-C2-H3 | -120.8838 | -120.8804 | - |
| N10-C1-C2-H4 | 0.0003 | 0.0001 | - |
| N10-C1-C2-H5 | 120.8843 | 120.8806 | - |
| C2-C1-C6-H7 | -0.004 | 0.0 | - |
| C2-C1-C6-H8 | 119.8733 | 120.0351 | - |
| C2-C1-C6-H9 | -119.8818 | -120.0351 | - |
| N10-C1-C6-H7 | 179.996 | -180 | - |
| N10-C1-C6-H8 | -60.1267 | -59.9649 | - |
| N10-C1-C6-H9 | 60.1182 | 59.9649 | - |
| C2-C1-N10-N11 | -180.0003 | -180.0001 | - |
| C6-C1-N10-N11 | -0.0002 | -0.0001 | - |
| C1-N10-N11-H12 | 0.0158 | -0.0009 | - |
| C1-N10-N11-C13 | -180.0046 | -180.0001 | - |
| N10-N11-C13-S14 | 180.0084 | -180.001 | - |
| N10-N11-C13-N15 | 0.0095 | -0.0015 | - |
| H12-N11-C13-S14 | -0.0108 | -0.0002 | - |
| H12-N11-C13-N15 | -180.0097 | -180.0007 | - |
| N11-C13-N15-H16 | 179.999 | -180.0037 | - |
| N11-C13-N15-H17 | -0.0031 | 0.0038 | - |
| S14-C13-N15-H16 | 0.0002 | -0.0042 | - |
| S14-C13-N15-H17 | -180.0019 | 180.0033 | - |
Table 1: Optimized bond parameters of Acetone Thiosemicarbazone.
The isotropic and anisotropic chemical shifts in 13C and 1H NMR to explore chemical environment of present compound were simulated in gas and solvent phase (DMSO and chloroform) under GIAO guidance at B3LYP/6-311G++(2d,p) level. Moreover the MEP view has been simulated using B3LYP/6-311++G(d,p) method and their depleted charge levels were represented by specific colors to explore that the reactive sites of the title molecule were evaluated. The thermodynamic functions (the heat capacity, entropy, enthalpy and Gibbs free energy) were calculated for the temperature region 100-1000K from the vibrational wavenumber calculations of the title compound. The VCD spectral analysis has been performed at B3LYP/6-311++G(d,p) level of calculation.
Molecular geometry deformation analysis
The present molecular crystal structure; Acetone-thiosemicarbazone fit in to CS group of symmetry and consists of 17 atoms within the molecule. The compound was made up of thiosemicarbazone chain adopted with couple of methyl group. The methyl groups making Y shape formation on imine group in thiosemicarbazone chain in which the different groups of atoms were found oriented in different planes. The crystal appearance of shaped compound in (111) pane was obtained in the Figure 1. According to the Cartesian coordinate, except methyl hydrogens, the entire molecular group was placed in one plane. In this case, thiosemicarbazone groups of atoms making semi hexagonal ring which causes the asymmetry of charge distribution. Also those groups were making non centro symmetry of the molecule which will lead the molecule fast optical response and was able to self-activate [20].

Figure 1: Molecular formation of Acetone thiosemicarbazone.
Two methyl groups on imine group were symmetrically placed in the molecule. But, according to the bond lengths C1-C2 (1.501 Ǻ) and C1-C6 (1.509 Ǻ), C6 group was 0.008 Ǻ stretched out than C1 due to the pulling of N adjacent to the that group. The observed bond length of N10-N11 (1.384 Ǻ) [21] was 0.020 Ǻ greater than calculated value (1.364 Ǻ) due to the C-N charge pulling. Though the imine was loaded on both sides by different group of atoms, there was no change observed in the bond length of C1=N11. This shows that the consistent of the imine group which was the reason for the NLO effect. The observed C-S bond length was 0.008 Ǻ lesser than the calculated value since the increment of the existed force constant. The bond angle C6-C1-N10 was to be the semi ring, but in this case, the methyl loading enhanced the bond angle 124º instead of 120º. The bond angle N10- N11-C13 was 6.04º greater than N11-C13-N15 in the semi hexagonal ring due to the substitution of S atom. This view showed the trouble of the semi hexagonal ring. The bond angle of C13-N15-H17 was exactly 120º which has not affected and making base moiety of the ring.
Mulliken charge distribution analysis
Usually, the design of organic molecular structure with equilibrium of charge distribution for the active response of optical activity is difficult. But, by adding the suitable ligands with the base, the desired charge distribution for the active optical response is easy. In this way, the thiosemicarbazone base was added with acetone and the preferred charge level arrangement was constructed. The thiosemicarbazone chain has specific bond length with imine and amine group of atoms which was fused with methyl twin. The Mulliken charge assignment was presented in the Table 2 and the related diagram was displayed in the Figure 2.
| Atoms | HF/6-311++G(d,p) | B3LYP/6-311++G(d,p) |
|---|---|---|
| 1C | 0.164291 | 0.008718 |
| 2C | -0.532651 | -0.489315 |
| 3H | 0.148793 | 0.150688 |
| 4H | 0.163002 | 0.16011 |
| 5H | 0.168023 | 0.170326 |
| 6C | -0.614225 | -0.541371 |
| 7H | 0.15346 | 0.158284 |
| 8H | 0.116856 | 0.115051 |
| 9H | 0.199714 | 0.195888 |
| 10N | 0.050244 | -0.030633 |
| 11N | -0.02426 | 0.14684 |
| 12H | 0.182117 | 0.147592 |
| 13C | -0.002273 | -0.195262 |
| 14S | -0.449985 | -0.31576 |
| 15N | -0.275397 | -0.18447 |
| 16H | 0.273727 | 0.246716 |
| 17H | 0.278564 | 0.2566 |
Table 2: Mulliken atomic charge distribution of Acetone Thiosemicarbazone.
| S No | Observed frequencies(cm-1) | Calculated frequencies(cm-1) | Species | Vibrational assignments | ||
|---|---|---|---|---|---|---|
| FT-IR | FT-Raman | HF/6-311++G(d,p) | B3LYP/6-311++G(d,p) | |||
| 1 | 3450vs | 3450vs | 3411 | 3445 | A′ | (N-H)υ |
| 2 | - | 3400vs | 3381 | 3416 | A′ | (N-H)υ |
| 3 | 3240s | 3240w | 3241 | 3235 | A′ | (N-H)υ |
| 4 | 3000m | 3000vs | 2990 | 2991 | A′ | (C-H)υ |
| 5 | 2970w | 2970s | 2979 | 2983 | A′ | (C-H)υ |
| 6 | - | 2960s | 2938 | 2959 | A′ | (C-H)υ |
| 7 | - | 2950s | 2931 | 2958 | A′ | (C-H)υ |
| 8 | - | 2940s | 2933 | 2938 | A′ | (C-H)υ |
| 9 | - | 2920s | 2930 | 2913 | A′ | (C-H)υ |
| 10 | 1560vs | 1560vs | 1565 | 1562 | A′ | (C=N)υ |
| 11 | 1520s | 1520vs | 1501 | 1518 | A′ | (C=S)υ |
| 12 | 1440m | 1440s | 1442 | 1448 | A′ | (N-H)δ |
| 13 | - | 1430w | 1431 | 1425 | A′ | (N-H)δ |
| 14 | - | 1410w | 1406 | 1404 | A′ | (N-H)δ |
| 15 | - | 1340s | 1355 | 1342 | A′ | (C-N)υ |
| 16 | 1260s | 1260s | 1248 | 1272 | A′ | (C-N)υ |
| 17 | 1250w | 1250s | 1232 | 1248 | A′ | (C-C)υ |
| 18 | - | 1190s | 1201 | 1188 | A′ | (C-C)υ |
| 19 | - | 1180s | 1198 | 1182 | A′ | (N-N)υ |
| 20 | 1150w | 1150m | 1146 | 1146 | A′ | (C-H)δ |
| 21 | 1110vs | 1110s | 1120 | 1102 | A′ | (C-H)δ |
| 22 | - | 1100s | 1105 | 1096 | A′ | (C-H)δ |
| 23 | 1070w | - | 1069 | 1068 | A′ | (C-H)δ |
| 24 | - | 1050m | 1060 | 1051 | A′ | (C-H)δ |
| 25 | 1040w | - | 1040 | 1039 | A′ | (C-H)δ |
| 26 | 870s | - | 862 | 866 | Aʺ | (N-H)γ |
| 27 | - | 850s | 851 | 856 | Aʺ | (N-H)γ |
| 28 | - | 840s | 845 | 839 | Aʺ | (N-H)γ |
| 29 | 780s | - | 779 | 777 | Aʺ | (C-H) γ |
| 30 | - | 770s | 763 | 769 | Aʺ | (C-H) γ |
| 31 | - | 760s | 750 | 759 | Aʺ | (C-H) γ |
| 32 | - | 690s | 689 | 681 | Aʺ | (C-H) γ |
| 33 | - | 620m | 609 | 613 | Aʺ | (C-H) γ |
| 34 | - | 610m | 610 | 599 | Aʺ | (C-H) γ |
| 35 | - | 550s | 553 | 545 | A′ | (C-N)δ |
| 36 | - | 520m | 522 | 536 | A′ | (C-N)δ |
| 37 | 480m | - | 483 | 479 | A′ | (C=N)δ |
| 38 | 410w | - | 404 | 413 | A′ | (C-C)δ |
| 39 | 340w | - | 338 | 340 | A′ | (C-C)δ |
| 40 | 300w | - | 300 | 289 | A′ | (N-N)δ |
| 41 | 280w | - | 278 | 284 | Aʺ | (C-N) γ |
| 42 | 240w | - | 247 | 230 | Aʺ | (C-N) γ |
| 43 | 210w | - | 212 | 210 | Aʺ | (C-C) γ |
| 44 | 180w | - | 184 | 174 | Aʺ | (C-C) γ |
| 45 | 110w | - | 108 | 104 | Aʺ | (N-N)γ |
Table 3: FT-IR and FT-Raman experimental and calculated (scaled) vibrational frequencies of Acetone Thiosemicarbazone.

Figure 2: Mulliken Charge Distribution in the compound.
In this present case, the highly positive atoms were represented by green color whereas highly negative atoms red color. Faded red color atoms showed less negative atoms and block color atoms symbolized neutral. Here thiocarbazone group was cemented with acetone molecule, thereby; the imine and azine groups were created in addition with thio group. Generally, in C=S and C=N groups, C and N and S would be positive and negative respectively and make the bond highly dipole character. But here, after the substitution of the CH3 groups in the compound, the C becomes highly negative and H turns out to be positive and thus the dipole with high degree was made in the methyl groups. Similarly, the N-H bonds become highly dipole. In the case of C=S, the positive level of C reduced much and the dipole character was faded. The Mulliken charge levels were drastically fluctuated in the corner atoms of the compound where as the core atoms were neutralized as much. The dipole moment of the entire compound was bringing out from such corner dipoles. After the substitution of CH3 group in the compound, the charge levels were redistributed and pulled by them. Except C1, the entire C was negative and N11 was less positive. The atoms in core joint were neutral to produce asymmetry of charge distribution among the atoms of the compound. This was the main basis of the present compound to be non centro symmetric and NLO active.
Vibrational assignments
The group frequencies from fundamental wavenumbers of Acetone thiosemicarbazone have been assigned according to their characteristic vibrational regions. The molecule belongs to CS point group symmetry that contains 17 atoms and it undergoes 45 normal vibrational modes. These active fundamental vibrations were distributed as 31 in plane vibrations denoted by A΄ species and 14 out of plane vibrations denoted by A˝ species, i.e., Γvib=31A΄+14A˝. The observed FT-IR and FT-Raman frequencies and calculated fundamentals at HF and DFT levels using the triple split valence basis set along with the diffuse and polarization functions; 6-311++G(d,p) have been presented in Table 3. And their respective spectra were displayed in the Figures 3 and 4. Comparison of calculated frequencies with the experimental values reveal the over estimation of the calculated vibrational modes due to the neglect of a harmonicity in real system. Inclusion of electron correlation in the density functional theory to certain extends makes the frequency values smaller in comparison with the HF data.

Figure 3: Experimental and calculated FT-IR spectra of Acetone Thiosemicarbazone.

Figure 4: Experimental and calculated FT-Raman spectra of Acetone Thiosemicarbazone.
Methyl group vibrations: In the present case, two methyl groups were present and it was most significant, because they are only the substitutional group in the compound. The fundamental vibrational modes of methyl groups are known to be influenced by the interactions such as electronic effects and intermolecular hydrogen bonding [22]. Electronic effects; back-donation and induction, mainly caused by the presence of C with the chain to CH3 group, can shift the position of C-H stretching and bending modes [23,24] to the lower region. The assignments of methyl group vibration make a significant contribution to the title molecule since it controls all other vibrations. The presence of C-H vibrations emphasized that, the place of methyl group in the chain. In CH3 substituted compounds, the C-H asymmetric and symmetric stretching vibrations appear in the range 2975-2840 cm-1 and 2870-2825 cm-1 respectively [25]. In the present case, the IR and Raman bands were at 3000, 2970, 2960, 2950, 2940 and 2920 cm-1 assigned to asymmetric C-H stretching vibrations. Though C-H was pure dipole bond, most of the bands were appeared in Raman spectrum instead of IR. These vibrational bands ensured the strong polarization existed in the compound. All these bands were observed at the top of the expected region which is due to the dominance character.
The C-H in-plane and out-of-plane bending vibrations for methyl substituted compounds are observed in the region of 1250-900 cm-1 and 650-900 cm-1 respectively [26-28]. Here, the C-H in-plane bending peaks were observed at 1150, 1110, 1100, 1070, 1050 and 1040 cm-1 and the out-of-plane bending vibrations identified at 780, 770, 760, 690, 620 and 610 cm-1 respectively. According to the reported literature [29,30], the entire in plane bending modes were found within the expected region whereas two of out of plane bending peaks were moved little behind the characteristic region. Hence, from this observation, it was clear that, this state of enhancement of CH3 vibrations in the vibrational pattern of the present compound ensured the existence of its physical and chemical property in the thiosemicarbazone.
N-H vibrations: Generally, N-H bonds in amine group always placed a consistent place in the entire vibrational pattern of the IR and Raman spectra due to their rich force constant. This is the uniqueness of the amine group which is usually dominates entire vibrational motion o the compound. Here, three N-H bonds acknowledged with the thiosemicarbazone group in the compound. The N-H bonds have to produce 9 vibrational modes and it will be the part of the whole vibrational pattern. Frequently, hetero nuclear bond N-H show its stretching vibration in the region 3500-3150 cm-1 [31]. The place of IR absorption of N-H vibration in the region depends upon the nature of hydrogen bonding and the substitutional effect [32]. In the present compound, thiosemicarbazone reserved three N-H bonds and the consequential stretching vibrations were found with very strong intensity in IR and Raman at 3450, 3400 and 3240 cm-1. The observed N-H stretching vibrations have not affected and also out of three vibrations, all were identified asymmetric. In this compound, except amino group, there were no dominated vibrations found. These stretching vibrations generally have not disturbed and this is true in this case.
The N-H bending (scissoring) vibrations usually observed in the region 1580-1640 cm-1 and the rocking modes are assigned in the range 1170-1080 cm-1 in thio amides which may be affected slightly by the inductive effect with high electronegative group [33-35]. Here, three in plane bending modes observed at 1440, 1430 and 1410 cm-1. Here, these vibrational modes should not be the scissoring bands since; the dominating vibrational bands should not be affected much. So, those bands were to be the rocking modes that were found encouraged above the expected region. This was mainly due to the thio group present in the compound which was also the root cause of the optical properties. The out of plane bending vibrations in thio amides are observed in the region 710-580 cm-1 [36]. Accordingly, the out of plane bending modes were identified at 870, 850 and 840 cm-1. Similar to the in plane bending, all the out of plane bands were elevated and prove its leading character. The source of second order optical response in crystal lattice was thio amide group and it was progressive in this compound.
C=N, C-N and N-N vibrations: The significant dipole bond (C=N and C-N) which was existed along with azine due to the substitution of methyl groups in the compound. Generally, the C=N stretching vibration has high force constant (13.43 mDyne/A) and high Raman activity (392.32 A4/AMU) observed in the region 1600-1490 cm-1 [37]. In this case, the vibrational band was found with very strong intensity at 1560 cm-1 in IR and Raman. This strong symptom of appearance the imine group ensured its very active presence. It’s in plane bending was present at 480 cm-1 which was also ensured the activity extension. Another important existence of dipole bond C-N and the resultant stretching absorption peak were observed in the region 1382–1266 cm-1 for amine derivatives [38]. In amide compound, the band observed at 1368 cm-1 was assigned to be due to C–N stretching [39]. Hence, in this case, two strong bands were observed at 1340 and 1260 cm-1 for C-N stretching vibrations. The in plane and out of plane bending vibrations are found at 550 and 520 cm-1 and 280 and 240 cm-1. The stretching vibrations have not affected whereas entire bending vibrations have suffered much due to the azine and thio group.
The rare electron-electron interaction bond N-N which is important core bond of azine group since it was bridge point for creating active NLO properties for title compound. Normally, the azine bond N-N stretching vibration was observed around 1080 cm-1 [40]. The N-N stretching vibration was strongly found at 1180 cm-1 in Raman spectrum only. This shows the higher optical activity of the molecule. The following in plane and out of plane bending modes was observed at 300 and 110 cm-1 respectively. The obtained azine group peaks were precisely harmonized with the literature and confirm its presence and explored the optical-semiconducting properties of the compound.
NMR profile: Nuclear magnetic resonance (NMR) spectroscopy has evolved as one of the most powerful analytical techniques for evaluating chemical properties of the organic compound. It permits to visualize single atoms and molecules in various media in solution as well as in solid state. NMR is nondestructive and gives molar response that allows structure elucidation and quantification simultaneously. The information provided by NMR spectroscopy regarding the structure of compounds has made this technique a great tool for the identification and characterization of base and ligand groups. When compared to other analytical techniques, NMR spectroscopy is one of the least sensitive techniques [41]. In this case, the optimized structure of present molecule is used to calculate the NMR spectra at B3LYP method with 6-311++G(d,p) level supported by GIAO method and the chemical shifts of the compound are reported in ppm relative to TMS for 1H and 13C NMR spectra which were presented in Table 4 and the corresponding spectra were shown in Figure 5.
| Atom | 1H NMR | Atom | 13C NMR | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Experimental | Theoretical (B3LYP) | Experimental | Theoretical (B3LYP) | ||||||
| Gas | DMSO | Chloroform | Gas | DMSO | Chloroform | ||||
| H12 | 9.9 | 7.82 | 7.98 | 7.93 | C13 | 181 | 185.8 | 184.68 | 185.18 |
| H17 | 8.6 | 6.58 | 7.03 | 6.90 | C1 | 151 | 146.96 | 157.34 | 154.52 |
| H16 | 7.9 | 5.41 | 5.71 | 5.63 | C2 | 40.3 | 26.45 | 26.63 | 26.58 |
| H5 | 4.5 | 1.98 | 2.17 | 2.11 | C6 | 17.6 | 13.88 | 14.41 | 14.24 |
| H3 | 3.3 | 1.98 | 2.17 | 2.11 | - | - | - | - | - |
| H4 | 2.5 | 1.81 | 1.77 | 1.79 | - | - | - | - | - |
| H8 | 1.9 | 1.72 | 1.84 | 1.81 | - | - | - | - | - |
| H9 | 1.8 | 1.72 | 1.84 | 1.81 | - | - | - | - | - |
| H7 | 1.8 | 1.64 | 1.88 | 1.81 | - | - | - | - | - |
Table 4: Experimental and calculated 1H and 13C NMR spectral data of Acetone Thiosemicarbazone.

Figure 5: Experimental and simulated 13C and 1H NMR spectra of Acetone Thiosemicarbazone.
The NMR signals are usually separated into different domain of peaks with respect the diamagnetic equivalence of carbon and hydrogen atoms. In this case, the carbons were appeared in three dissimilar environments such as methyl C, amine C and imine C. The chemical shift of C13 was more shifted than C1, C2 and C6 since its shield was completely broken by the loading of N and S atoms unevenly. The observed isotropic chemical shift of C1 was 141 ppm and it was high when compared with C2 and C6. This was mainly due to the breaking of paramagnetic shield by methyl and imine groups. The chemical shift of C2 and C6 were very low and below 50 ppm which was due to the further shielding of delocalized electrons. It was also evidenced (negative carbons) in the Mulliken charge analysis. The chemical shift of hydrogens in amine group was high when compared with methyl hydrogens which was mainly due to the coupling of N. The chemical shift of methyl hydrogens found to be very low due to the highly positive flavor occurrence. The entire hydrogen atoms in this compound were found with high degree of positive flavor which was due to making intensive dipole with corresponding atoms. Thus, entire hydrogens in the compound were participated in the alternation of the chemical properties of the compound.
Frontier molecular analysis- electronic properties: The frontier molecular orbitals are very much useful for studying the electric, linear and non linear optical properties of the organic compound. The stabilization of the molecular- bonding orbital and destabilization of the antibonding orbital is achieved by the overlapping of two orbitals [42]. In the case of molecular orbital interaction, two important molecular orbitals called frontier orbitals (HOMO and LUMO) interact with each other. After the formation of molecular orbital, generally, the electron density will occupy at the region between two nuclei. In the molecular orbital, the in-phase interaction (bonding orbital) and out of phase interaction (anti bonding molecular orbital) is taking place in ligand and base molecule [43].
The 3D diagram of the frontier molecular orbitals, HOMO and LUMO for title compound were displayed in Figure 6. From the figure, it was noted that, the HOMO was mainly localized over the π-orbital lobe of carbon and sulphur coupling of thio group and the region upon which the electron cloud with intense density that was common for both C and S. from this condition, it was inferred that, the thio group acting as donor for N-H, C-N bonding interaction. The σ-bond lobe interactions were identified N-N, C-N and N-H bonds which also acted as donor for the interaction. There was spatial extension formed up in the C=S π-delocalization. In LUMO, there were so many empty interactive orbitals appeared in S, C-N, C-C, C-H, N-N and N-H which were able to create σ-orbital lobe interactions. The σ- bonding interactions were identified in different bonds of LUMO and also some of the extreme cascade orbital interactions appeared in the core bonds. The σ- electrons interactions taking place in between thio group (donor) and methyl groups (acceptor) in which several lobe were interacted with one another and making as chain orbitals. This was the main reason for the reducing the energy gap of the compound. The calculated electronic energy gap was found to be 3.54 eV which was in the range of semiconducting property. In addition to that, in the case of subsequent orbitals in LUMO-1, the σ-bond lobe interaction was made together to form umbrella shape spatial quantization of orbitals and it was clearly seen in the Figure 6.

Figure 6: Frontier molecular orbital in IR region.
Optical properties analysis: Generally, UV/Vis spectroscopic tool is used in analytical chemistry for the quantitative determination of different bonded elements, such as highly conjugated molecular system in base compound. UV-Visible spectroscopy is useful in the structure elucidation of organic molecules related to chromophores and to study the optical property the molecule and also to detect the NLO activity of the compound. The calculations of the electronic structure and electronic energy excitation were calculated at the B3LYP/6-311++G(d,p) level using the TD-DFT approach in gas phase and with the solvent of DMSO and Chloroform. The calculated excitation energies, oscillator strength (f) and wavelength (λ) and spectral assignments are given in Table 5. The TD-DFT calculations predicted that, irrespective of the gas and solvent phase, the entire transitions belong to quartz ultraviolet region. In the case of gas phase, the strong transition was observed at 254.58 nm with maximum oscillator strength f=0.23 with 4.87 eV energy gap. The entire transitions were assigned to n→σ* and n→π*. The designation of the band was R (German, radikalartig) and attributed to the chromophores groups, such as C=N, C=S and N-N group. The electronic transitions belong to hypsochromic shift and the solvent effect was found to be less in this compound. The Frontier molecular orbital interaction in UV-Visible region was depicted in the Figure 7. According to the figure, the σ and π-bond delocalization taken part in the interaction which had given the stabilized energy gap 4.94 eV. This was so high and enough to strong stabilization between optical donor and optical acceptor of the compound. When the LUMO moved to very lower levels, the umbrella molecular interaction was observed which helped to stimulate optical polarization. The experimental UVVisible spectrum was found to be same as calculated spectrum which was presented in the Figure 8.

Figure 7: Frontier molecular orbital in UV-Visible region.

Figure 8: UV-Visible spectra of Acetone Thiosemicarbazone.
| λ (nm) | E (eV) | ( f) | Major contribution | Assignment | Region | Bands |
|---|---|---|---|---|---|---|
| Gas | ||||||
| 340.5 | 3.64 | 0.0056 | H®L (91%) | n→σ* | Quartz UV | R-band (German, radikalartig) |
| 254.5 | 4.87 | 0.2301 | H-1®L (81%) | n→π* | ||
| 244.3 | 5.07 | 0.0363 | H®L+1 (88%) | n→π* | ||
| DMSO | ||||||
| 334.1 | 3.71 | 0.0126 | H®L (83%) | n→σ* | Quartz UV | R-band (German, radikalartig) |
| 264.3 | 4.69 | 0.3054 | H-1®L (79%) | n→π* | ||
| 238.8 | 5.19 | 0.0141 | H-2®L (57%), H-3®L (40%) | n→π* | ||
| Chloroform | ||||||
| 336.1 | 3.69 | 0.0111 | H®L (86%) | n→σ* | Quartz UV | R-band (German, radikalartig) |
| 262.9 | 4.72 | 0.3169 | H-1®L (82%) | n→π* | ||
| 236.3 | 5.25 | 0.0117 | H-3®L (48%), H-2®L (47%) | n→π* | ||
Table 5: Theoretical electronic absorption spectra of Acetone Thiosemicarbazone using TD-DFT/B3LYP/6-311++G(d,p) method.
In UV-Visible spectra, entire transitions belong to the quarts ultraviolet region (240-340 nm). So the compound also can be used for quarts optics [44]. The consistent peak was observed at 300 nm in both experimental and calculated spectrum which was due to the presence of two π conjugated system of bonds (C=N, C=S) in the compound. From this observation, it was clear that, the present compound is able to produce the optical energy with second and third fold frequency. When it was going from gas to solvent phase, it was determined that, the wavelength region was decreased and simultaneously the energy of the absorption was increased. This was the important evidence of increment of frequency in the present compound.
Molecular electrostatic potential (MEP) maps: The Molecular electrostatic potential view is simulated by the B3LYP/6-311+G(d,p) level of theory and was presented in the Figure 9. The MEP is highly informative concerning the protonic and electronic charge distribution of a given molecule. It is mainly used for biological interactions analysis similar to the crystalline state. It is also used for topographical analysis of the electronic structure and molecular reactivity pattern of the complex molecules.

Figure 9: MEP display of Acetone Thiosemicarbazone.
The organic structure with large delocalized π-systems have proven to be more easily affected by an external optical field as they are relatively loosely bound to the nucleus and that the delocalized orbitals may be extended over the entire molecule giving large and fast polarization [45]. The color code of these maps was represented between -6.75 a.u. (genuine red) to 6.75 a.u. (genuine blue). Generally, the negative regions are mainly localized over the portion of highly electronegative atoms. The Figure 9 showed the two extremities of polarized charges in which, the nucleophilic region was appeared wherever the hydrogens present in the compound whereas the electrophilic region was found only on sulphur of thio group. The charge depletion region was identified at the core bonds of the compound. The possibility of the charge separation was taking place strongly between thio and CH3 groups. This was making resultant strong dipole arrangement in this present compound which will help to the improvement of NLO activity.
NLO activity analysis: To design of efficient NLO compound, in order to increase the nonlinearity, it was important that, the donor acceptor capability in the base compound is to be increased by the substitutions attached to the π-conjugated system. The position of the substitutions is of vital important in terms of NLO activity. The large value of the first hyperpolarizability, which is the measure of the nonlinear optical activity of the molecular system, is associated with intramolecular charge transfer resulting from an electron cloud movement through a π conjugated framework from electron donor to electron acceptor groups.
In this case, the acetone as functional group was added with thiosemicarbazide base, the present compound was capitulated. The calculated parameters were presented in the Table 6. The polarizability (α) and hyper polarizability (β0) of the thiosemicarbazide were 63 × 10-33 esu and 142 × 10-33 esu respectively whereas the same for acetone were 40 × 10-33 esu and 91 × 10-33 esu respectively. But the polarizability (α) and hyper polarizability (β0) of the present compound were 160 × 10-33 esu and 285 × 10-33 esu. From this observation it was clear that, the second order and third polarization were streamlined. Thus, the substitutional effect of acetone has extensively improved the nonlinearity flavor of the thiosemicarbazone. In addition to that, the position of CH3 making Y shape structure and was of vital important in terms of NLO activity. The stabilization of the energy between HOMO and LUMO is depends upon the polarization existed in the compound while it absorb the electrical field. This can be identified by measuring the dipole moment. If the compound possesses high degree of dipole moment, normally, the compound will be responding effectively for the flow of local electric field and suppress the wavelength and multiply the frequency. Here, the dipole moment was found to be 5.71 Debye and it was high enough to induce non-linear characteristics.
| Parameters | B3LYP/6-311++G(d,p) | ||
|---|---|---|---|
| μx | 5.574 | ||
| μy | 1.255 | ||
| μz | -0.0001 | ||
| μ | 5.71 (Debye) | ||
| Δα | 220.39 | ||
| αTotal | 160.72 | ||
| βxxx | 61.905 | ||
| βxxy | 15.499 | ||
| βxyy | -2.259 | ||
| βyyy | 18.994 | ||
| βxxz | -0.0003 | ||
| βxyz | 0.0006 | ||
| βyyz | -0.0007 | ||
| βxzz | 7.112 | ||
| βyzz | -0.471 | ||
| βzzz | 0.0000 | ||
| βtotal | 285.718 × 10-33 (esu) | ||
| Parameters | Acetone | Thiosemicarbazide | Acetone thiosemicarbazone |
| Dipole moment(μ) (Debye) | 2.9664 | 4.0548 | 5.713 |
| Average Polarizabilities(α0) | 64.206 | 59.652 | 220.393 |
| Exact polarizability (Δα)×10-33esu | 40.753 | 63.016 | 160.727 |
| Hyperpolarizability (β0) ×10-33esu | 91.329 | 142.517 | 285.718 |
Table 6: Calculated electric dipole moments μ (Debye), dipole moment components, α and βtot components of Acetone thiosemicarbazone.
NBO profile: The natural bond orbital (NBO) calculations were made on natural bonding and antibonding orbital transitions. The calculations has been performed along with the frequency calculation using Gaussian NBO 6.0 program [46] with B3LYP/6-311++G(d,p) method, in order to determine electronic transitions from the filled orbital of one bond system and unfilled antibonding orbital of another system. The NBO profile of the compound was used for the quantity of energy transition, type of delocalization of electron density and kind of hyper conjugation of electronic orbitals. The NBO analysis granted the information regarding electronic orientations inside the molecule after the renovation of molecular orbitals after the optimized structure was formed.
The Natural Bond Orbitals parameters; occupancy, the energy difference between donors and acceptors, stabilization energy, polarization energy etc, were listed in Table 7. The allowed transitions between various possible donors and acceptors in molecule with their occupancy value were presented in detail. According to the selection rule, the stabilization energy for these transitions gives a measure of the likelihood of these transitions which indicate the highly probable in this molecule. The transition was observed from C1–N10 to N11– C13 (σ–σ* with 10.54 Kcal/mol). This transition indicated that, the energy was transferred between the consecutive bonds of the chain. The transition taking place from C1–N10 to C6-H8 (π-σ*, 10.42 Kcal/mol) which showed that, the energy transformation from hyper conjugation to single electron conjugation system. The exchange of energy was from N10-N11 to C1-C2 and C13-S14 (σ -σ*, 14.4 and 9.6 Kcal/mol) which was to be the important energy transfer since the N-N bond is the vital responsibility for NLO property of the compound.
| Type | Donor (i) | Occupancy ED/e | Acceptor (j) | ED/e | E(2) KJ/mol | E(2) kcal/mol | Energy difference E(j)-E(i) a.u. | Polarized energy F(i,j) a.u. |
|---|---|---|---|---|---|---|---|---|
| σ -σ* | C1 - C2 | 1.97808 | C1 - N10 | 0.01912 | 7.32 | 1.75 | 1.25 | 0.04 |
| N10 - N11 | 0.03491 | 24.81 | 5.93 | 1.01 | 0.07 | |||
| σ -σ* | C1 - C6 | 1.98765 | C1 - N10 | 0.01912 | 5.77 | 1.38 | 1.27 | 0.04 |
| σ -σ* | C1 - N10 | 1.98856 | C1 - C2 | 0.01904 | 4.77 | 1.14 | 1.33 | 0.04 |
| C1 - C6 | 0.03486 | 4.94 | 1.18 | 1.31 | 0.04 | |||
| N11 - C13 | 0.0586 | 10.54 | 2.52 | 1.35 | 0.05 | |||
| π -σ* | C1 - N10 | 1.95195 | C2 - H3 | 0.01018 | 10.42 | 2.49 | 0.73 | 0.04 |
| C2 - H5 | 0.01018 | 10.42 | 2.49 | 0.73 | 0.04 | |||
| C6 - H8 | 0.00908 | 7.7 | 1.84 | 0.72 | 0.03 | |||
| C6 - H9 | 0.00908 | 7.7 | 1.84 | 0.72 | 0.03 | |||
| σ -σ* | C2 - H3 | 1.97476 | C1 - N10 | 0.01912 | 7.11 | 1.7 | 1.12 | 0.04 |
| C1 - N10 | 0.18809 | 18.07 | 4.32 | 0.53 | 0.04 | |||
| σ -σ* | C2 - H4 | 1.98763 | C1 - C6 | 0.03486 | 19.2 | 4.59 | 0.91 | 0.06 |
| σ -σ* | C2 - H5 | 1.97476 | C1 - N10 | 0.01912 | 7.11 | 1.7 | 1.12 | 0.04 |
| C1 - N10 | 0.18809 | 18.07 | 4.32 | 0.53 | 0.04 | |||
| σ -σ* | C6 - H7 | 1.98763 | C1 - N10 | 0.01912 | 22.43 | 5.36 | 1.13 | 0.07 |
| σ -σ* | C6 - H8 | 1.97596 | C1 - C2 | 0.01904 | 7.91 | 1.89 | 0.94 | 0.04 |
| C1 - N10 | 0.18809 | 17.32 | 4.14 | 0.54 | 0.04 | |||
| σ -σ* | C6 - H9 | 1.97596 | C1 - C2 | 0.01904 | 7.91 | 1.89 | 0.94 | 0.04 |
| C1 - N10 | 0.18809 | 17.32 | 4.14 | 0.54 | 0.04 | |||
| σ -σ* | N10 - N11 | 1.98467 | C1 - C2 | 0.01904 | 14.43 | 3.45 | 1.27 | 0.06 |
| C13 - S14 | 0.01241 | 9.62 | 2.3 | 1.17 | 0.05 | |||
| σ -σ* | N11 - H12 | 1.98721 | C13 - N15 | 0.04985 | 15.52 | 3.71 | 1.15 | 0.06 |
| σ -σ* | N11 - C13 | 1.98836 | C1 - N10 | 0.01912 | 7.57 | 1.81 | 1.46 | 0.05 |
| N15 - H16 | 0.00802 | 9.25 | 2.21 | 1.27 | 0.05 | |||
| σ -σ* | C13 - S14 | 1.98217 | C13 - S14 | 0.49924 | 24.39 | 5.83 | 0.21 | 0.04 |
| π -σ* | C13 - S14 | 1.9764 | N10 - N11 | 0.03491 | 19.16 | 4.58 | 1.05 | 0.06 |
| C13 - N15 | 0.04985 | 4.48 | 1.07 | 1.16 | 0.03 | |||
| N15 - H17 | 0.01951 | 13.31 | 3.18 | 1.1 | 0.05 | |||
| σ -σ* | C13 - N15 | 1.99177 | N11 - H12 | 0.03538 | 9.96 | 2.38 | 1.24 | 0.05 |
| σ -σ* | N15 - H16 | 1.98839 | N11 - C13 | 0.0586 | 18.41 | 4.4 | 1.1 | 0.06 |
| σ -σ* | N15 - H17 | 1.98528 | N11 - C13 | 0.0586 | 4.39 | 1.05 | 1.11 | 0.03 |
| C13 - S14 | 0.01241 | 23.1 | 5.52 | 0.98 | 0.07 | |||
| n -σ* | LPN10 | 1.91979 | C1 - C2 | 0.01904 | 7.49 | 1.79 | 0.81 | 0.03 |
| C1 - C6 | 0.03486 | 46.74 | 11.17 | 0.79 | 0.09 | |||
| n -σ* | LPN10 | 1.91979 | N11 - H12 | 0.03538 | 38.53 | 9.21 | 0.77 | 0.08 |
| N15 - H17 | 0.01951 | 6.61 | 1.58 | 0.81 | 0.03 | |||
| n -π* | LPN11 | 1.66979 | C1 - N10 | 0.18809 | 114.1 | 27.27 | 0.29 | 0.08 |
| C13 - S14 | 0.49924 | 273.93 | 65.47 | 0.22 | 0.11 | |||
| n -σ* | LPS14 | 1.98468 | N11 - C13 | 0.0586 | 11.3 | 2.7 | 1.12 | 0.05 |
| C13 - N15 | 0.04985 | 15.27 | 3.65 | 1.16 | 0.06 | |||
| n -σ* | LPS14 | 1.88099 | N11 - C13 | 0.0586 | 47.2 | 11.28 | 0.62 | 0.08 |
| C13 - N15 | 0.04985 | 48.87 | 11.68 | 0.66 | 0.08 | |||
| n -σ* | LPN15 | 1.71823 | C13 - S14 | 0.49924 | 320.83 | 76.68 | 0.21 | 0.12 |
Table 7: NBO analytical data of Acetone Thiosemicarbazone.
The N11-C13 to C1-N10 and N15-H16 (σ-σ*, 7.5 and 9.2 Kcal/ mol) which showed the maximum possibility of energy exchange and causing the pathway of lobe interaction. This was evidenced in HOMO LUMO energy operation. The energy switching was taking place even from LP N10 to C1-C2 and C1-C6 (σ-σ* with 7.4 and 46.4 Kcal/mol). This transition was rare and found in this compound which tells the strong stabilization arranged in those orbitals. These transitions with high magnitude showed the energy stabilization of the chain. Some of the σ-σ* transitions were taking place between base and substitutions which indicated that, the strong interaction were occurred and leaded to the enhancing of NLO properties of substitutions with the thiosemicarbazone.
Thermodynamical functions analysis: The thermodynamic functions; entropy, specific heat capacity and enthalpy of the crystal compound play a significant process in physical as well as chemical properties. The thermodynamical calculation was performed using NIST program with suitable method; B3LYP/6-311++G (d,p) and was represented in the Table 8 and the corresponding diagram was depicted in the Figure 10. The coefficients of entropy, specific heat capacity and enthalpy are always varied with respect to temperature positively in the case of semiconducting compound. In this case, when the temperature increased from 100K to 1000K, the thermodynamical functions found to vary as linear curve and sustained up to the maximum temperature. This linear variation shows the present compound was being semiconductor.
| T (K) |  |
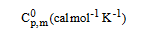 |
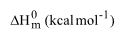 |
Gibbs free energy 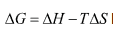 KJmol-1 KJmol-1 |
|---|---|---|---|---|
| 100 | 318.20 | 120.76 | 7.20 | -31812.8 |
| 200 | 428.65 | 204.68 | 23.61 | -85706.4 |
| 298.15 | 523.81 | 275.66 | 47.24 | -15612.7 |
| 300 | 525.52 | 276.95 | 47.75 | -157608.0 |
| 400 | 614.41 | 342.99 | 78.81 | -245685.0 |
| 500 | 697.29 | 400.34 | 116.05 | -348529.0 |
| 600 | 774.65 | 448.33 | 158.56 | -464631.0 |
| 700 | 846.86 | 488.37 | 205.45 | -592597.0 |
| 800 | 914.34 | 522.18 | 256.03 | -731216.0 |
| 900 | 977.56 | 551.08 | 309.73 | -879494.0 |
| 1000 | 1036.94 | 576.00 | 366.11 | -1036574.0 |
Table 8: Thermodynamic properties at different temperatures.

Figure 10: Thermodynamic variation diagram of Acetone Thiosemicarbazone.
Similarly, the Gibbs free energy is positive temperature coefficient as well. This positive temperature coefficient of the compound proved to be the optical semiconductor. Normally, the chemical reaction is feasibly occurring when the Gibbs free energy of the molecular system decreases as much as possible. Here, the Gibbs free energy ΔG was found to be negative which revealed that, the chemical reaction was constant and it was inferred that, the present compound was chemically strong and active. The thermodynamic functions are normally reflect the chemical viability of the compound and showed the ability to bind with another ligand groups in order to extend their optical property. Here, the variation of those functions exposed that; the optical property can be tuned further by suitable substituents.
Chemical properties: The physical property of the organic compound is directly related with chemical property. So, the physical parameters are very important to clarify the chemical behavior of the compound. Accordingly, the chemical hardness and potential, electronegativity and Electrophilicity index were calculated and presented in Table 9. Molecules with polar covalent bonds can result in molecules with overall polar attributes. The measure of the overall polarity of a molecule is called the dipole moment. As the value of the dipole moment increases, so does the polarity of the molecule. In this case, the dipole moment reflects linear as well as non linear optical property of the compound which is the capability of electrical and optical polarization of a compound. The large dipole moment leads the compound; very strong intermolecular interactions. The calculated dipole moment value for the present compound was 5.71 Debye and it was comparatively very high enough to induce strong intermolecular interactions. This strong interactive force is able to create second order polarization in the compound.
| Parameters | Values | ||
|---|---|---|---|
| HOMO | -8.24218 eV | ||
| LUMO | -4.69835 eV | ||
| Energy gap | 3.54383 eV | ||
| Ionization potential (IP) | -8.24218 eV | ||
| Electron affinity (EA) | -4.69835 eV | ||
| Electrophilicity Index (ω) | 11.812 | ||
| Chemical Potential (μ) | 6.470 | ||
| Electronegativity (χ) | -6.470 eV | ||
| Hardness (η) | -1.772 | ||
| Softness (S) | 0.2822 | ||
| Dipole moment (Debye) | 5.713 Debye | ||
| Parameters | Thiosemicarbazide (A) | Acetone (B) | Electrophilicity charge transfer (ECT) (ΔNmax)A-(ΔNmax)B |
| Chemical Potential (μ) | 6.012 | 3.82 | +0.47 |
| Chemical Hardness (η) | -3.438 | -2.99 | |
| ΔNmax= -μ/ η | 1.75 | 1.28 | |
Table 9: Chemical parameters of Acetone thiosemicarbazone.
The chemical hardness is a significant factor which was used to estimate the crystal hardness and thermodynamic stability of organic compound. The chemical hardness was found to be 1.772 which was perceptible magnitude by which the present compound has definite hardness. Here, hardness character was enriched by adding thiosemicarbazone with acetone group. The tendency for an atom to attract electrons is its electronegativity. Usually, the rate of electronegativity is depends upon the functional groups of the compound. Here, the compound was composed by two separate groups of atoms and electronegativity was found to be 6.470, it was relatively high and local polarization was effectively present. Also the property of chemical bonds of title molecule was being ionic.
Electrophilicity index is a factor which is used to measure the energy lowering due to maximal electron flow between ionization potential and electron affinity. The Electrophilicity index was 11.81 and was high which ensured the energy transition between donor (HOMO) and acceptor (LUMO). This was inferred that, the band gap of the present compound was within the limit and exhibit the semiconducting nature. The electrophilicity based charge transfer (ECT) is another important physico chemical function which provides the exchange of energy and charges from base to functional group of vice versa. If ECT is greater than zero, the charges will be moved from base to functional group. If ECT is less than zero, the charges will be tending to move from functional group to base compound. In this case, the base and functional group were thiosemicarbazone and was acetone respectively. The ECT was +0.47 which was greater than zero. So, here, the charges moved from acetone to thiosemicarbazone to persuade the NLO property.
VCD analysis: Vibrational circular dichroism (VCD) is electrooptic effect which exhibits vibrational optical activity of IR form and detects differences in attenuation of left and right circularly polarized light passing through the compound. It is the extension of circular dichroism spectroscopy into the IR and near infrared region. The VCD provides two-dimensional vibrational structural information since VCD is sensitive to the mutual orientation of base and ligand groups in a molecule. It is also used for the identification of absolute configurations of the organic compounds. The result of a VCD measurement is the combination of emission and absorption spectra, figure out the elucidation of chemically and optically significant molecules.
The VCD spectrum and (Mirror reflection) enantiomers of the present compound was portrayed in Figure 11. In this present case, though, the spectrum was initiated from zero, the rigorous signals were appeared in mid infrared region and the intensity of the absorption and emission were elevated in positive and negative dimensions due to the left and right circular polarization. The absorption intensity is usually unequal on both sides (up and down) of the VCD spectrum, in this case, the probability of incorporation was found to be higher in absorption side than emission. This was mainly due to the C=N, C=S, C-C, N-N, N-H and C-N stretching vibrations. There were two additional spectral peaks observed for N-N and C-H stretching. In the present case, the optical second and third order dichroism was effective and this circumstance was optimistic to the temptation of the NLO properties.

Figure 11: VCD spectra of Acetone Thiosemicarbazone.
The geometry of the present molecule was optimized in HF and DFT level of theories and the optimized parameters have been calculated and presented. The alternation of the bonds and bond angles with respect to methyl groups are discussed. The Mulliken charge analysis has been carried out in order to explain the redistribution of charges and simultaneously, the change of physical and chemical properties are interpreted. From the observation, it was clear that, the molecule is very reactive for electrical and optical properties. As this compound optically active, the addition of acetone group was further improving the electronic and optical activity. The vibrational sequence of the molecular bonds corresponding to the region of IR and Raman regions are studied in detail. From the NMR chemical examination, it is very important to note that, the introduction of acetone groups as new substituent in a compound was increased the crystal efficiency in moderate way. From the optical results, it is observed that, the molecular Polarizability and hyperpolarizability are dynamic. So, the present compound can be used to prepare NLO crystals. From the NBO analysis of the present compound, it is believed that, most of the interactions are taking place between BD to BD* and BD to RY*. Thus, by the electron cloud transformation, the charge distribution is customized according to the orbital interaction.