Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2016) Volume 5, Issue 1
Background: Statins exert immune effects and have been shown to strongly inhibit the interferon-γ induced expression of the human leukocyte antigen (HLA) DR. Monocyte HLA-DR is decreased severe in sepsis and indicates hypo inflammatory and immunosuppressive phases. The aim of the study was to investigate the statin-induced regulation of constitutive HLA-DR expression on monocytes.
Method: Monocytes and the monocyte cell line Mono Mac 6 were incubated with simvastatin, mevastatin, pravastatin and fluvastatin. HMG-Coenzyme A reductase was inhibited by l-mevalonate. Protein expression of HLA-DR, Major histocompatibility complex, invariant CD74 and apoptosis were measured by flow-cytometry. mRNA expression was assessed by PCR.
Results: Simvastatin dose dependently reduced the constitutive HLA-DR expression on monocytes starting from 500 nM. Bypassing statin-induced inhibition of HMG- Coenzyme A reductase reversed this effect. HLA-DR was also decreased in response to mevastatin, pravastatin, lovastatin and fluvastatin after 24 h. Simvastatin induced caspase-3 activation and DNA-fragmentation in a small subpopulation of Mono Mac 6 cells. HLA-DR expression was lower on apoptotic cells, but simvastatin also reduced HLA-DR on non-apoptotic cells. Simvastatin did no repress HLA-DR mRNA. For transferring HLADR to endosomes the chaperon CD74 is required. Intracellular CD74 was decreased by simvastatin and fluvastatin.
Conclusion: Statins decrease the constitutive expression of MHC II on monocytes and Mono Mac 6 independently of apoptosis. Reduced intracellular levels of the chaperon CD74 by statins may potentially impede HLA-DR surface expression.
Keywords: HLA-DR; MHC II; Statin; Sepsis; CD74; Monocyte
Evidence from large observational studies indicated that statins may have benefits in the prevention of sepsis [1], acute respiratory distress syndrome [2] of patients with cardiovascular diseases [3] or kidney failure [4], as well as in ICU patients [5]. In animal models statins increase survival administered prior to and even after the experimental induction of sepsis [6]. Instead, a recent meta analysis of randomized clinical trials failed to reveal a survival benefit of de novo statin therapy in sepsis and acute respiratory distress syndrome [7,8]. Statin use prior to sepsis however improved survival of sepsis [9].
Statins are inhibitors of 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMG-CoA reductase) [10]. Apart from their mere cholesterol-lowering activity they display pleiotropic immune effects [10], for example they inhibit the interaction of antigen presenting cells and T-cells [11] and induce apoptosis in several cell types [12,13]. In endothelial cells, however, they inhibit apoptosis [14].
The interaction of antigen presenting cells and T-cells is a crucial step in the activation of the adaptive immune system and depends on the presentation of the target antigen by the major histocompatibility complex II (MHC II) [11]. The human leukocyte antigen (HLA) DR is a part of MHC II. The transport of HLA-DR from the Golgi system to endosomes, where antigen loading takes place, depends on the presence of its chaperone CD74 (invariant chain) [15,16].
Statins have been shown to strongly inhibit the interferon-γ induced expression of HLA-DR, a main component of MHC II [11]. However, in severe sepsis due to a shift from TH1 to TH2 T-cell profile [17], interferon-γ concentrations are relatively low [17-19]. Furthermore, HLA-DR expression is downregulated in severe sepsis [20,21]. A long lasting low HLA-DR expression (6 days) correlated with sepsis severity scores and death [22]. HLA-DR in severe sepsis is down regulated on a transcriptional level [22]. Low HLA-DR is a predictor of increased mortality [23,24]. However, sepsis scores showed a better discriminatory power in the prediction of mortality [25].
The aims of this study were to investigate, (1) if different statins also inhibit the constitutive expression of HLA-DR without prior activation by interferon-γ, (2) whether this effect could be attributed to accelerated apoptosis, and whether (3) statins also affect the expression of the HLA-DR chaperone CD74.
Cell culture
Heparinized blood (Sarstedt, Frankfurt, Germany) was obtained from healthy human volunteers after informed consent with approval of the local ethics committee. The mononuclear fraction was obtained using a Ficoll gradient (Sigma-Aldrich, Steinheim, Germany). Monocytes were isolated by negative magnetic bead separation according to manufacturer’s recommendations (IMag Human Monocyte Enrichment Set, BD Biosciences, Heidelberg, Germany). Purity of monocytes after separation was monitored using flow cytometry. Monocytes were identified by double positive staining for CD14 (allophycocyanin labelled, clone M5E2, BD Biosciences, Heidelberg, Germany) and CD33 (fluorescein isothyocyanate labelled, clone HIM3-4, BD Biosciences, Heidelberg, Germany) resulting in a purity of 78% to 85%. After washing in phosphate buffered saline (PBS, Sigma-Aldrich, Steinheim, Germany), cells were placed in cell culture medium (90% RPMI 1640+10% FBS+2 mM L-glutamine (Invitrogen [GIBCO], Karlsruhe, Germany) with 100 U/mL penicillin and 100 μg/ ml streptomycin at 37°C with 5% CO2) at a density of 1 × 106 for further treatment. The Mono Mac 6 cell line (Leibnitz Institute DSMZ-German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany) [26] was used as a model of monocytes to further study statin induced regulation of HLA-DR. Cultures were monitored to exclude mycoplasma contamination according to manufacturer’s instructions (Venor GeM Mycoplasma Detection Kit, Sigma-Aldrich, Steinheim, Germany). Cells were cultured in 90% RPMI 1640+10% FBS+2 mM L-glutamine+non-essential amino acids+1 mM sodium pyruvate+9 μg/ml bovine insulin (Invitrogen [GIBCO], Karlsruhe, Germany) at 0.3-1.0 × 106 cells/ml in 24 well-plates (Greiner-bio-one, Solingen, Germany) at 37°C with 5% CO2. For experiments passages 25-45 were used. The identity of the cell line was authenticated by surface staining and flowcytometric detection. The cell line expressed CD14 (BD Biosciences, Heidelberg, Germany) but not CD3 (CD3-12 AlexaFluor 488, AbD Serotec, Puchheim, Germany). For experiments, cells were placed in 24 well plates in fresh medium at a density of 1.0 × 106 cells/mL.
Cell treatments
Simvastatin, mevastatin, pravastatin, lovastatin and fluvastatin were obtained from Merck, (Darmstadt, Germany). Simvastatin and mevastatin were solved in a stock solution (20 nM) in ethanol, while the other statins were prepared in deionised water. Cells were incubated with statins in concentrations of 0.1-20 μM for 24 hours in a cell culture incubator at 37°C in an atmosphere containing 5% CO2. To bypass the inhibition of the 3-hydroxy-3-methyl-glutaryl-Co-enzyme A reductase (HMG-CoA reductase) L-mevalonate (100 μM, Sigma Aldrich, Steinheim, Germany) was added prior to statin treatment. This concentration of L-mevalonate is sufficient to prevent the atorvastatininduced depletion of cholesterol [27].
Apoptosis was induced by incubation with the activating monoclonal antibody anti-Fas IgM (clone CH11, Immunotech, Marseille, France) at a final concentration of 200 ng/ml containing protein G for 8 hours or by staurosporine (2 μM for 4 hours). In certain experiments lipopolysaccharide (LPS, 2 ng/mL, Escherichia coli 055:B5, Sigma Aldrich, Steinheim, Germany) or interferon-γ (IFN-γ, 400 U/ml Biomol, Hamburg, Germany) were used as controls.
Quantification of protein expression
Staining of surface proteins was carried out essentially as published earlier [21] and adapted for cell culture. 5 × 105 cells were incubated with phycoerythrin (PE) or fluorescein isothyocyanate (FITC) labelled antibodies against a non-polymorphic epitope of HLA-DR (MHC class II, clone L243, BD Biosciences, Heidelberg, Germany), HLA ABC (MHC class I, clone G 46-2.6, BD Biosciences, Heidelberg, Germany) and CD74 (clone M-B741, BD Biosciences, Heidelberg, Germany) for 30 minutes at room temperature after which the samples were fixed with FACS-Lysis-Solution (Becton Dickinson, Heidelberg, Germany), washed twice and stored in the dark at 4°C until analysis.
For intracellular staining of proteins cells were permeabilized after blocking surface bound antigens with unlabelled antibodies against HLA-DR (clone L243, BD Biosciences, Heidelberg, Germany) or CD74 (clone M-B741, BD Biosciences, Heidelberg, Germany) respectively for one hour at 4°C. Cells were washed with PBS, (Sigma- Aldrich, Steinheim, Germany) and fixed with 750 μl PBS containing 4% paraformaldehyde (Sigma-Aldrich, Steinheim, Germany). After fixation, cells were permeabilized with 1ml PBS containing 0.5% Saponin (Sigma-Aldrich, Steinheim, Germany) and 1% bovine serum albumin (Sigma-Aldrich, Steinheim, Germany). Then, cells were incubated with PE-labeled antibodies against HLA-DR or CD74 in PBS containing 0.5% saponin/1% BSA, followed by washing.
Detection of apoptosis
The activation of caspase-3 was assessed by intracellular staining of the active fragment and evaluation by flow cytometry as described [28]. For mono-stained cells a PE-labeled antibody and for dual-staining a FITC- labeled antibody was used. DNA-fragmentation was monitored as the subdiploid DNA peak using flow cytometry according to Nicoletti et al. [29] modified as described [28].
Flow cytometry
Ten thousand events of interest were acquired by flow cytometry (FACSCalibur, Becton Dickinson, Heidelberg, Germany) and analyzed with CellQuest Pro software (CellQuest Pro, Version 4.0.2, Becton Dickinson, Heidelberg, Germany). Isotype controls were used for exclusion of background fluorescence. Mean fluorescence intensity (MFI) expressed as linear fluorescence units (LFU) and percentage of positive cells were recorded.
mRNA expression
RNA-extraction, cDNA synthesis and quantification of gene expression by real-time PCR were carried out essentially as described [30]. PCR reactions were performed under the following conditions: initial denaturation at 95°C for 15 minutes followed by 45 amplification cycles: 95°C for 15 seconds, 55°C for 35 seconds, 72°C for 30 seconds followed by an additional heating to 95°C for melting curve detection. Hypoxanthine phosphoribosyltransferase-1 (HPRT-1) was used as a housekeeping gene. It had been selected by the laboratory previously for the study of human leukocytes in the context of sepsis [30,31]. Also, in comparative studies HRPT-1 has been found suitable for gene expression studies in peripheral blood of septic patients [32]. It has also been selected for the study of HLADR on monocytes [33]. For specific genes the following primers were purchased: HPRT 5’-TGACCTTGATTTATTTTGCATACC, 5’-CGAGCAAGACGTTCAGTCCT (Operon, Cologne, Germany); HLA-DR α-chain Refseq No. NM_019111.3 Band Size: 97 bp Reference Positions: 712-733 (SuperArray, Frederick, MD, USA). For quantification the crossing point method was employed. Concentrations were calculated as “normalized ratio” with Relative Quantification Software (Roche, Mannheim, Germany).
Statistics
ANOVA analysis was applied to evaluate differences between different treatments. For post hoc testing the Bonferroni test was employed, when appropriate. Values were given as mean +/- SEM (standard error of the mean). Analysis was done with GraphPad for Windows (Version 3.02, San Diego, CA, USA). Experiments were carried out in duplicates and were reproduced at least twice. Significance was accepted at p<0.05.
Human monocytes were exposed to varying concentrations of simvastatin and the expression of HLA-DR was recorded by flow cytometry (Figure 1a). Starting at a concentration of 500 nM simvastatin reduced the surface expression of HLA-DR (p<0.05) with a maximal effect of 28% reduction at 20 μM (p<0.01, Figure 1b). However, simvastatin did not influence the expression of MHC class I on Mono Mac 6 cells (ethanol control 765 ± 14 vs. 704 ± 26.5 LFU).
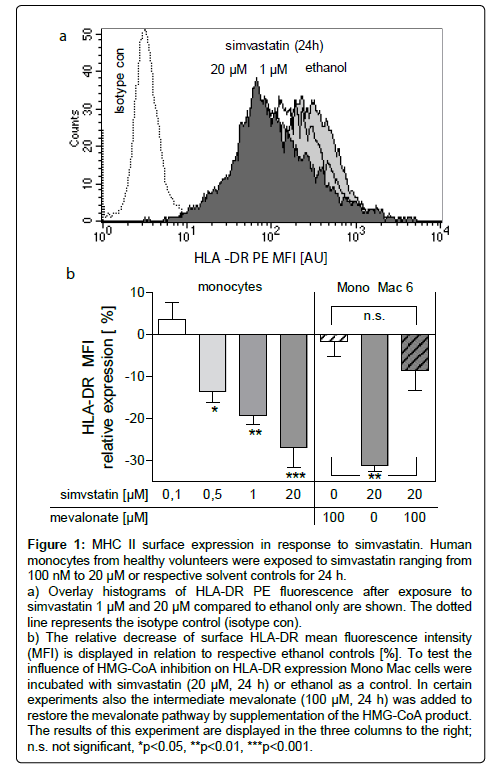
Figure 1: MHC II surface expression in response to simvastatin. Human monocytes from healthy volunteers were exposed to simvastatin ranging from 100 nM to 20 μM or respective solvent controls for 24 h.
a) Overlay histograms of HLA-DR PE fluorescence after exposure to simvastatin 1 μM and 20 μM compared to ethanol only are shown. The dotted line represents the isotype control (isotype con).
b) The relative decrease of surface HLA-DR mean fluorescence intensity (MFI) is displayed in relation to respective ethanol controls [%]. To test the influence of HMG-CoA inhibition on HLA-DR expression Mono Mac cells were incubated with simvastatin (20 μM, 24 h) or ethanol as a control. In certain experiments also the intermediate mevalonate (100 μM, 24 h) was added to restore the mevalonate pathway by supplementation of the HMG-CoA product. The results of this experiment are displayed in the three columns to the right; n.s. not significant, *p<0.05, **p<0.01, ***p<0.001.
The observed effect was not restricted to simvastatin. The influence of simvastatin, mevastatin, pravastatin, lovastatin and fluvastatin on HLA-DR expression was tested in the Mono Mac 6 cell culture model at a concentration of 20 μM. All tested statins significantly lowered the expression of HLA-DR, simvastatin and fluvastatin showing the highest potencies (Table 1). Down-regulation was compared to respective solvent controls (ethanol for simvastatin and mevastatin, H2O for the others).
| Statin | ∆ MFI [%] +/- SEM | p |
| Mevastatin | -7.27+/-0.66 | <0.05 |
| Pravastatin | -14.07+/-1.44 | <0.01 |
| Lovastatin | -14.39+/-1.22 | <0.01 |
| Simvastatin | -26.98+/-2.12 | <0.001 |
| Fluvastatin | -29.50+/-3.09 | <0.001 |
Table 1: Modulation of HLA-DR expression by different statins.
To investigate whether statins inhibit HLA-DR expression in an HMG-CoA reductase dependent way, mevalonate was used to bypass statin-induced inhibition of HMG-CoA reductase. Mevalonate (100 μM, 24 h) did not downregulate HLA-DR but it reversed the statininduced inhibition of HLA-DR expression on Mono Mac 6 cells (p<0.01, Figure 1b).
Simvastatin (20 μM, 24 h) consistently induced apoptosis only in a small proportion of Mono Mac 6 cells. It activated caspase-3 in a small proportion of cells (4.38 ± 0.02% vs. solvent control 2.33 ± 0.1% p<0.01) and caused a small but statistically significant increase in DNA-fragmentation (7.88 ± 0.23% vs. 6.1 ± 0.15%, p<0.05). Induction of apoptosis using an activating antibody to Fas or staurosporine caused a reduction of HLA-DR expression in cells with active caspase-3 (p<0.001, Figure 2a). Thus, if apoptosis induction per se decreases HLA-DR expression, simvastatin potentially could reduce HLA-DR via a mechanism of apoptosis induction. Therefore, Mono Mac 6 cells were double stained for HLA-DR and active caspase-3 after incubation with simvastatin (20 μM for 24 h). In this model simvastatin decreased HLA-DR expression both in non-apoptotic cells (p<0.001) and cells positive for caspase-3 (p<0.001 vs. ethanol control, not significant vs. active caspase-3 negative population, Figure 2a). Staining for lowered DNA-content (sub-G1 population) was used to detect late apoptotic cells. In response to simvastatin (20 μM, 24 h) HLA-DR was decreased in the subpopulation with normal DNA-content (p<0.001), but was significantly lower in cells of the sub G1 population (p<0.001, Figure 2b). When apoptosis was induced with an activating antibody against Fas or staurosporine, reduction of HLA-DR was observed in the apoptotic cell populations. In the absence of statin in cells with intact DNA, HLA-DR expression was not affected, while it decreased in the sub G1 population after incubation with aFas (p<0.001, Figures 2a and 2b).
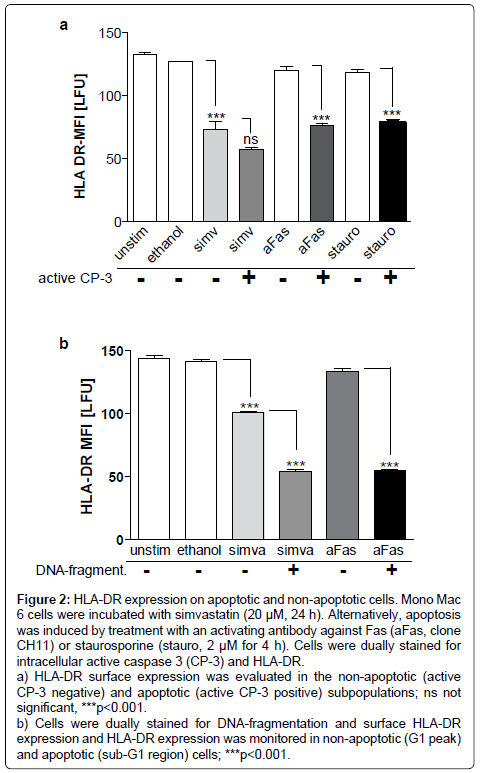
Figure 2: HLA-DR expression on apoptotic and non-apoptotic cells. Mono Mac 6 cells were incubated with simvastatin (20 μM, 24 h). Alternatively, apoptosis was induced by treatment with an activating antibody against Fas (aFas, clone CH11) or staurosporine (stauro, 2 μM for 4 h). Cells were dually stained for intracellular active caspase 3 (CP-3) and HLA-DR.
a) HLA-DR surface expression was evaluated in the non-apoptotic (active CP-3 negative) and apoptotic (active CP-3 positive) subpopulations; ns not significant, ***p<0.001.
b) Cells were dually stained for DNA-fragmentation and surface HLA-DR expression and HLA-DR expression was monitored in non-apoptotic (G1 peak) and apoptotic (sub-G1 region) cells; ***p<0.001.
To investigate whether lowered HLA-DR expression in response to simvastatin is regulated by inhibition of respective mRNA-expression, we quantified HLA-DR α-chain mRNA. However, Simvastatin did not change HLA-DR α-chain mRNA-expression after incubation for 3 h (1208 ± 156.6 AU; solvent control 1305 ± 502.3), 12h (1303 ± 443 AU), or 24 h (1250 ± 449.4 AU), while a positive control with interferon-γ massively increased mRNA expression (7268 ± 1699 AU, p<0.001). Correspondingly, we evaluated the intracellular HLADR protein content. In contrast to the findings regarding surface expression, intracellular HLA-DR content did not decrease in response to simvastatin but showed a tendency to increase (65.55 ± 0.46 vs. 56.45 ± 2.40, p<0.05).
Intracellular HLA-DR is sorted from the golgi apparatus to late endosomes in the presence of its chaperone CD74. This step is necessary for later transfer of HLA-DR to the cell surface. Surface CD74 expression was not affected by simvastatin (97.48% + -9.2% vs. solvent control 98.7 ± 1.99% relative MFI). Regarding intracellular CD74 protein content in Mono Mac 6 cells, we observed that simvastatin as well as fluvastatin dose-dependently decreased the expression of CD74 (p<0.001 vs. ethanol control, Figure 3). 20 μM simvastatin was as effective as LPS 2 ng/ml in Mono Mac 6 cells. As a positive control, interferon-γ induced CD74 expression.
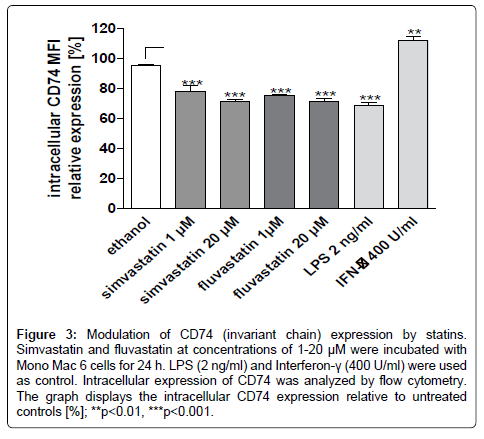
Figure 3: Modulation of CD74 (invariant chain) expression by statins. Simvastatin and fluvastatin at concentrations of 1-20 μM were incubated with Mono Mac 6 cells for 24 h. LPS (2 ng/ml) and Interferon-γ (400 U/ml) were used as control. Intracellular expression of CD74 was analyzed by flow cytometry. The graph displays the intracellular CD74 expression relative to untreated controls [%]; **p<0.01, ***p<0.001.
The study shows that the constitutive HLA-DR expression on monocytes and Mono Mac 6 cells is downregulated by statins. This process does not depend on apoptosis induction by statins. Furthermore, statins also inhibit the intracellular expression of CD74, the invariant chain.
Initially, statins have been shown to strongly inhibit the interferon-γ mediated induction of HLA-DR on macrophages and endothelial cells [11], while the constitutive expression of MHC II on dendritic cells was not affected by atorvastatin [11]. In phytohemagglutinin-stimulated Tand B-cells simvastatin repressed MHC II without prior interferon-γ treatment. In sepsis, due to a shift from TH1 to TH2 cytokine profile, interferon-γ concentrationsare relatively low [17-19]. Therefore, analysis of constitutive MHC II expression in resting monocytes may better reflect the situation in sepsis than after interferon-γ stimulation. Our studies indicate that also the constitutive MHC II expression on resting monocytes and Mono Mac 6 cells is repressed by statins. The extent of inhibition is comparable to the effect of simvastatin observed in unstimulated dendritic cells in another study [34].
Significant repression of surface HLA-DR in our experiments was observed starting at a concentration of 500 nM. In human liver microsomes the half maximal inhibitory concentrations (IC50) for simvastatin regarding HMG-CoA reductase activity is 100-300 nM [35]. In studies downregulation of interferon-gamma induced HLADR by atorvastatin [11] and induction of macrophage apoptosis by lovastatin [13] were observed at statin concentration of 10 μM. In endothelial cells simvastatin inhibited apoptosis at 1 μM. Thus, the statin concentrations causing HLA-DR reduction observed in the current study are in accordance with other described immune effects of statins.
Thus, the effect on HLA-DR appears at physiologically relevant concentrations. Statins exhibit several effects independent of HMGCoA inhibition such as inhibition of P-glycoprotein [36]. The observed effect in our study however could be attributed to the inhibition of HMG-CoA reductase since bypassing HMG-CoA reductase by addition of mevalonate reversed the action of statin.
In our experimental setting simvastatin and fluvastatin were the most efficient statins in regard to decrease in HLA-DR surface expression followed by pravastatin, lovastatin and mevastatin. While statins are taken up actively in liver cells, in other cell types passive diffusion across membranes occurs [37]. Fluvastatin and simvastatin are more lipophilic than pravastatin which results for example in a lower potency of pravastatin to inhibit vascular smooth muscle proliferation [37]. Similarly, differences in lipophilicity may account for the different potencies of statins to repress constitutive HLA-DR expression in our study.
Apart from other anti-inflammatory actions, statins have been demonstrated to induce apoptosis in RAW264.7 macrophages [12], murine bone marrow cells [13] and resting T-cells [38]. Mechanisms included activation of the Rac1/Cdc42/JNK pathway [13] and an increase of the Bax/Bcl-2 ratio [38]. In our model system Mono Mac 6 simvastatin induced apoptosis only in a small subpopulation of cells. We were able to demonstrate that induction of apoptosis by activation of the receptor mediated extrinsic pathway or by staurosporine also decreases HLA-DR surface expression. Therefore, statins in principle could decrease HLA-DR already by induction of apoptosis. However, dual staining in flow cytometry revealed that HLA-DR is clearly downregulated by simvastatin in non-apoptotic cells. Thus, the MHC II lowering action of statins cannot only be attributed to induction of apoptosis in our model but occurs independently of apoptosis. Yet, in other cell types, where statins induce apoptosis more potently, this mechanism could become more important and may distort findings if experiments are not controlled for apoptosis.
The transcription of HLA-DR and other proteins of the MHC II group is controlled by the class II transactivator (CIITA) [39]. In interferon-γ treated human vascular endothelial cells statins inhibited HLA-DR expression on a transcriptional level via decreased expression of CIITA and its promoter pIV-CTA [11]. Yet, in U937 monocytes, HeLa cervical carcinoma cells and human activated primary T-cells the statin effect on interferon-γ induced MHC II regulation is regulated on a post-transcriptional level [39]. Due to the fact that the mevalonate pathway produces both cholesterol and isoprenoids, posttranscriptional activities of statins include disruption of cholesterol microdomains [40] and inhibition of small G-proteins that need isoprenoid modifications for proper targeting within the cell [41]. In unstimulated Mono Mac 6 cells we did not observe inhibition of HLA-DR α-chain mRNA expression by simvastatin. Therefore, posttranscriptional mechanisms are likely to be involved.
In the endoplasmatic reticulum (ER) MHC II heterodimers associate with the invariant chain CD74, which assists egress from the ER and directs MHC II complexes into the endocytic pathway where further processing and antigen loading take place [15]. Mice lacking invariant chain expression show a marked reduction in surface MHC class II expression [42]. While interferon-γ upregulates CD74, LPSpriming reduces intracellular CD74, which has been shown to inhibit HLA-DR expression on monocytes in response [43]. In Mono Mac 6 cells we could show that both simvastatin and fluvastatin decreased intracellular CD74. Reduced availability of the invariant chain in response to statin treatment may contribute to reduced HLA-DR surface expression by impeding HLA-DR transport. This finding may reflect the results of a clinical study where CD74 gene expression increased and correlated with HLA-DR expression during the recovery phase from septic shock [44]. Decreased CD74 expression in whole blood predicted mortality after septic shock [45]. In further studies the mechanism of statin-induced CD74 regulation and its effect on HLADR expression should be explored.
Evidence points towards a protective effect of statin use prior to sepsis [2]. However, it is still a matter of debate whether inhibition of HLA-DR expression by statins is beneficial in sepsis [46], since monocytes are deactivated in sepsis by reduction of HLA-DR [20,21,47], and since low HLA-DR expression during sepsis is associated with increased mortality [23,24]. During sepsis and LPStolerance, HLA-DR is downregulated on a transcriptional level [48,49]. Thus, post-transcriptional effects by statins could result in additive HLA-DR decrease. Large observational studies have shown a sepsispreventing effect of statin intake [3,4]. Potentially, attenuation of the initial pro-inflammatory phase in early sepsis by chronic HLADR downregulation may mitigate the ensuing dangerous immune dysregulation. On the other hand downregulation of monocyte HLADR with ensuing monocyte deactivation may contribute to failure of statins in the treatment of sepsis, [7,8] when started after the onset of sepsis.
In conclusion, statins reduce the constitutive expression of HLADR on monocytes via a mechanism that does not depend on apoptosis induction but may be associated with reduced expression of the chaperon invariant chain CD74. Further studies should analyze the statin-induced down regulation of the invariant chain CD74 in the context of HLA-DR downregulation in patients with sepsis.
The authors would like to thank Caroline von dem Bussche for excellent technical assistance. The authors declare that they have no conflict of interest. The work was supported by intramural funding from BONFOR (Bonn University research grant organisation).