Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2010) Volume 3, Issue 6
Next generation sequencing has revolutionized the status of biological research. For a long time, the gold standard of DNA sequencing was considered to be the Sanger method. However, in 2005, commercial launching of next generation sequencing has made it possible to generate massively parallel and high resolution DNA sequence data. Its usefulness in various genomic applications such as genome-wide detection of SNPs, DNA methylation profi ling, mRNA expression profi ling, whole-genome re-sequencing and so on are now well recognized. There are several platforms for generating next generation sequencing (NGS) data which we briefl y discuss in this mini overview. With new technologies come new challenges for the data analysts. This mini review attempts to present a collection of selected topics in the current development of statistical methods dealing with these novel data types. We believe that knowing the advances and bottlenecks of this technology will help the researchers to benchmark the analytical tools dealing with these data and will pave the path for its proper application into clinical diagnostics.
Keywords: DNA; Sequencing; Deep sequencing; High throughput; Sequence reads; RNA; ChIP-seq; Intensities
Next generation sequencing (Shendure and Ji, 2008), also known as deep sequencing, is a transformative technology for today’s biomedical research. The growing importance of next generation sequencing for the clear understanding of various biological systems has indirectly triggered a competition among several companies, each trying to come up with a sequencing platform which can produce high quality longer read sequences with greater throughput and reduced cost. We begin this mini review by discussing various technologies for next-generation sequencing.
Roche 454
NGS using Roche 454 technology became commercially available in 2005 (Margulies et al., 2005). This technology uses bead-based emulsion polymerase chain reaction (em- PCR) to amplify copies of templates of DNA molecule (Dressman et al., 2003). The amplified beads are sequenced in parallel by pyrosequencing (Marsh, 2007). In pyrosequencing, four different nucleotides are flowed in a sequential manner through a solid surface containing wells into which single beads can fit. This process goes on for cycles and the signal intensity per flowing nucleotide is recorded for each bead over time and is analyzed to generate good quality sequence.
This platform is lot more high throughput than any capillary based sequencing. In the Titanium version of the Roche 454 platform, the output has several hundred mega bases of 400-500 base reads per run. Hence, it is more cost effective than Sanger’s chain termination method (Sanger et al., 1977) which was the old standard of DNA sequencing. The 454 technology does not suffer from the G-C rich content and does not skip the unclonable segments as the process does not rely on cloning. However, it is to be noted that 454 technology suffers from detecting subsequences of repetitive DNA sequences or homopolymers in a DNA sequence. As pyrosequencing depends on intensities of light, the light emitted for detecting TAAAA or AAAAA could be very similar. Also, while 454 sequencing is cheaper and faster per base, each run is quite expensive (over $8000), and so it is not suitable for sequencing targeted fragments from small numbers of DNA samples.
SOLiD by applied biosystem
In this technology, similar to 454, DNA fragments are amplified by em-PCR onto beads (Dressman et al., 2003). The difference between the SOLiD and the 454 platform is that the SOLiD beads are much smaller than 454 beads (1 μm vs. 28 μm ). This results in much denser packing of beads into the same area in SoLiD (100 million beads per sequencing run). This platform can produce approximately 20 Gb of short-read sequence data per run (25-50 bases) and so is preferable to resequencing for de novo assembly. SOLiD uses a unique ligationmediated sequencing strategy which is less prone to the errors involved with pyrosequencing method in 454 platform. In the SOLiD system, each data point represents two adjacent bases, and each base is interrogated twice. Hence it can discriminate between sequencing errors and true polymorphisms. The drawback of this platform is that the data is collected in color space and it provides information about two adjacent bases but not the definitive identification. So, they have to be decoded in order to be mapped to a reference genome and conventional alignment tools can’t be used for the mapping process. Direct conversion from color to sequence data also is prone to error such as reads that contain sequencing errors can not be converted accurately. Error rate of this platform is significantly higher than traditional Sanger sequencing.
Illumina/solexa genome analyzer
This platform was first introduced by Solexa in 2006 and later on re-branded as Illumina Genome Analyzer (GA). This technology does not depend on the em-PCR to amplify the template DNA strands likethe other two platforms mentioned before. Instead, adapter ligated template molecules flow into the flow cell (hollow glass slide). Template DNA hybridizes to the primers on the flow cell surface and gets copied onto the flow cell as an extension to the hybridization primer. This results in a generation of reverse complimentary copy of the template. This newly synthesized strand serves as templates for isothermal amplification reaction and results in clusters of amplified strands. Due to the terminator nucleotides, each DNA strand within a cluster incorporates the same nucleotide within a cycle. The clusters are imaged and the next round of nucleotide incorporation begins after removing the imaged blocked groups and the flurophores of the newly incorporated nucleotides. Analysis of the images generates a separate sequence for each cluster.
An Illumina Genome Analyzer is currently capable of producing a sequence up to 10-Gb per 76-cycle paired-end run. However, beyond this length the frequency of the substitution errors are high due to cluster phasing and de-phasing.
Helicos true single-molecule-sequencing (tSMS) technology
This sequencing platform has been launched in 2008, and is considered as the next-next generation or 3rd generation sequencing. This platform is based on the technology that was published in 2003 (Braslavsky et al., 2003). This sequencing also deals with millions of templates parallely; however, tSMS differs from the existing next generation sequencing described earlier in that it does not amplify the template molecules. So it is free from any errors due to the amplification process. Also, the library preparation is simple and rapid. In this technology, single molecules are the substrates for the sequencing reaction. Fluorescent nucleotides are added singly. The flow cell is visualized to identify strands that hybridized the particular nucleotide with the help of the fluorescence of the nucleotide. The incorporation of the nucleotides to the strand depends on the compatibility of the template strand with the order of the nucleotide addition. Hence the length of the sequences is variable but on average they are of lengths of 25-30 bases.
This technology is free from phasing errors. However, sensitivity seems to be an issue with this platform (Harris et al., 2008). The true error rate can be reduced from 2 to 3% to below 1% by repeating the reading of the same strand twice. However, it increases the running time (Harris et al., 2008).
Other than these major platforms, some noteworthy emerging platforms are mentioned here. The SMRT technology by Pacific Biosciences (Eid et al., 2009) has recently showed promising early results using single-molecule real-time DNA sequencing. Dover Systems’ Polonator was announced in early 2008 by George Church at MIT and arose from collaboration between George Church’s laboratory and Danaher Corporation. Although- this system uses bead based em-PCR (Dressman et al., 2003) and sequencing by ligation (Shendure et al., 2005), this is very high throughput and can generate data up to 3 Gb per day. However, read lengths are short and so it is difficult to use them for vertebrate-size genome. An appealing feature of this technology is that it is open source and the users can buy all the reagents from any supplier. Other NGS platforms include: BASE (single-molecule sequencing technology) by Oxford Nanopore Technologies/Illumina, one by Intelligent Bio-systems using proprietary sequencing-bysynthesis technology (Ju et al., 2006) and single-molecule sequencing technology based on fluorescence resonance energy transfer (FRET) by VisiGen Biotechnologies etc. Sequencing technologies are also under development by Affymetrix Reveo, Base4innovation, Genome Corp, and Complete Genomics, among others. Detailed technical reviews of various NGS platform appeared in Mardis (2008) and Metzker (2010).
A brief comparison of the three most popular NGS platforms
Roche 454 gives longer reads (500-700 bp) than both Illumina and SOLiD but it suffers from low accuracy in the long homopolymeric regions.
The price for sequencing each nucleotide is several times reduced in the Illumina technology compared to the Roche 454 pyrosequencing platform. In terms of the total analysis time and sequencing throughput, Illumina and SOLiD platforms are close to each other (flow cell construction being costlier in Illumina and sequencing in SOLiD).
From the perspective of practical applications, both Illumina and SOLiD platforms have their respective cutting edges. In highthroughput resequencing of large genomes SOLiD is more accurate than Illumina whereas for RNA sequence analysis (RNA-seq), Illumina is more suitable.
Base calling techniques
Most of the work in this important research area has taken place primarily for the llumina (Solexa) platform. Its base calling can suffer from three dominant noise factors (Erlich et al., 2008) as follows. In sequencing-by-synthesis, each single-stranded nucleotide fragment is amplified around the initial attachment in the flow cell, resulting in a cluster of about 1,000 identical copies of each fragment. Eachterminal nucleotide in all the clusters is then excited by lasers and its signal is detected by charged coupled device (CCD) images of fluorescence emission. Ideally, the current position for synthesizing will be the same within a cluster, generating a strong signal (Figure 1a). However, the unstable chemistry causes stochastic failures in reading the next nucleotide, introducing phasing (lagging; no new base synthesized) and prephasing (leading; two bases synthesized) noises (Figure 1b). The second noise factor is due to loss of copies of fragments so that the signal intensity is reduced, which is called the fading noise (Figure 1c). The third noise factor is known as the fluorophore cross-talk, causing misinterpretation of the signal (Figure 1d).

Figure 1: a: The Illumina noise factors. Here the identical DNA templates are depicted as the colored boxes and the black ovals denote DNA polymerases. With no noise, the current signal (red lightning) is strong enough to infer the DNA sequences directly. b: When the phasing noise is introduced, it causes lagging (green lightning) or leading (blue lightning). c: The loss of material can cause reducing the signal intensity (fading noise). d: The fl uorophore crosstalk can cause misinterpretation of the current signal.
Illumina developed the built-in base-caller Bustard to transform observed intensities into sequences. Bustard consists of three steps and each step deals with the three main noise factors separately. It first handles the fluorophore cross-talk by transforming intensities to concentrations. To do this, it defines the cross-talk matrix and removes the overlapping fluorophore effect from intensities by taking the inverse crosstalk matrix. Next renormalization of concentrations is performed by dividing by the average concentration to eliminate the fading noise. The third step involves fitting a Markov model to eliminate the phasing noise resulting in the estimated sequences.
Rougemont et al. (2008) used probabilistic modeling and modelbased clustering to identify and code ambiguous bases and to arrive at decisions to remove uncertain bases towards the ends of the reads. Alta-Cyclic was developed by Erlich et al. (2008) based on support vector machine (SVM), requiring a control lane containing a sample with a known reference genome for supervised learning. Another attempt to improve the Illumina basecaller led to Swift by Whiteford et al. (2009). They devoted it to the image analysis.
One of the primary challenges in base calling is the dependency among cycles. Bustard, including Alta-Cyclic, assumes that all the cycles are performed independently. Recently, several cycledependent base-callers have been introduced. Ibis (Improved base identification system) was developed based on the SVM by Kircher et al. (2009). They used the multiclass-SVM to provide for a cycledependent model differently from Alta- Cyclic in which univariate SVM was used (Erlich et al., 2008). Bravo and Irizarry (2009) came up with their own modeling to quantify the read/base-cycle effects. Recently, Kao et al. (2009) developed BayesCall based on a stochastic Bayesian modeling. A somewhat complex dynamic modeling strategy is used in BayesCall which is schematically described in Figure 2, where L refers to the total number of cycles (length of fragments) in a run, Sk= (S1,k, S2,k,…SL,k) represents the complementary DNA sequence with length L in cluster k, It,k =(It,kA ,It,kC ,It,kG ,It,kT )′€R4×1 denotes the observed fluorescence intensities of the A, C, G, T channels at cycle t in cluster k, and εt,k denotes the active template concentration in cluster k at the t-th cycle. One of the novelties of BayesCall is the capability to use cycle-dependent parameters in its modeling, adding greater flexibility. To avoid over-fitting, the read length is divided into nonoverlapping windows and it is assumed that the parameters remain constant within each window. In general, three types of algorithms are used to estimate the parameters in BayesCall, namely, MCEM (Wei and Tanner, 1990), ECM (Meng and Rubin, 1993) and simulated annealing (Kirkpatrick et al., 1983). Finally, a quality score for a call is calculated based on its estimated posterior probability. For further details we refer the readers to the original paper by Kao et al. (2009).

Figure 2: The graphical modeling for BayesCall when the window size is 1.
For the Roche (454 Life Sciences) platform, there exist two base callers that are the built-in 454 base caller and Pyrobayes (Quinlan et al., 2008). The Applied Biosystems (SOLiD) uses a different style to detect the signal by the two base color code and there currently is only its own built-in base-caller.
Data quality and reproducibility
Several papers have examined the reliability and reproducibility of data from next generation sequencing platforms. While some studies have found next generation sequencing data to be superior to competing methods, others have found systematic problems with the reads obtained in next generation sequencing. Most of these studies used data obtained from the Illumina platform.
Marioni et al. (2008) observed that next generation sequencing data from Illumina are highly reproducible and very reliable, and overall they found it to be superior to the data produced by microarray technology. They used Illumina to sequence each sample on seven lanes across two plates. The gene counts were highly correlated across lanes (Spearman correlation average = 0.96).
To test for a lane effect by comparing each pair of lanes, Marioni et al. (2008) tested the null hypothesis that gene counts in one lane represent a random sample from the reads in both lanes for each mapped gene. Let, for a sample t, xtk denote the observed number of counts in lane k and let Ck denote the number of reads in lane k for k = a, b. For a clear understanding of this test for a lane effect, it is helpful to let Xtk denote the random variable representing the number of counts in lane a in an experiment with xta+xtb total counts, Ca reads from lane a, and Cb reads from lane b. Now, they tested the null hypothesis that the gene counts in one lane represent a random sample from the reads in both lanes for each mapped gene; symbolically, this is a test of the null hypothesis
H0 : π = P0 (xta) versus the alternative HA : π ≠ P0 (xta) where π = P (Xta= xta) and
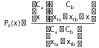
is the probability that Xta =x if the null hypothesis is true (in which case Xta follows a hypergeometric distribution). They used this test to compute the P-values for each gene and plotted the quantiles of the uniform distribution against the observed quantiles of the P-values for each gene, and they found that less than 0.5% of the genes had small P-values when the pair of lanes had the same concentration of samples. However, a larger proportion of genes indicated a laneeffect when the pair of lanes had different concentrations.
Marioni et al. (2008) also suggested a global test for lane effects by comparing all L lanes. For each sample i, they assume that the number of reads mapped to gene j for lane k follows independent Poisson distributions with mean cik λijk where cik is the total rate that lane k produces reads at and λijk is the rate of reads to gene j in lane k relative to other genes. To test the null hypothesis
H0: λij1 =…= λijL (i.e., λijk are equal for each lane k = 1,…, L ) versus the alternativeHA:λijk are not equal for all k, they used a goodness-of-fit statistic which follows a chi-square distribution with L -1 degrees of freedom when the null hypothesis is true. After plotting the Chi-squared quantiles against the observed quantiles for each gene j, they found that only about 0.5% of the genes had extra Poisson variation when lanes sequenced the same sample at the same concentrations.
Marioni et al. (2008) also used the Poisson model to identify differentially expressed genes. Specifically, for each gene j, they tested the null hypothesis that the rate of reads λijk are the same for all i and k versus the alternative that the liver and kidney sample have different read rates
λijkA. and λijkB Here, they used the likelihood ratio test statistic which follows a X12 -distribution under the null hypothesis. Using this method, 11493 genes were found to be differentially expressed in the liver-versus-kidney samples. This list of differentially expressed genes obtained with the Illumina data was compared with the results based on Affymetrix U133 Plus 2 arrays where an empirical Bayes approach was used to identify differentially expressed genes. Of 8113 differentially expressed genes found by the array, 81% were also found to be differentially expressed using Illumina. Finally, quantitative PCR (qPCR) was used to examine discrepancies, and overall, the qPCR results agreed more with Illumina than with the arrays.
Fu et al. (2009) arrived at a similar conclusion by comparing the relative accuracy of transcriptome sequencing (RNA-seq) and microarrays with protein expression data from adult human cerebellum using 2D-LC MS/MS. They found that the next generation sequencing provided more accurate estimation of absolute transcript levels.
Wall et al. (2009) used simulation models to compare next generation sequencing with traditional capillary-based sequencing and concluded that next generation sequencing offers great benefi t in terms of coverage over capillary-based sequencing. However, they suggest combining sequencing methodologies such as FLX and Solexa to achieve optimal performance at a modest cost.
On the other hand, a number of authors have reported problems and systematic biases with the sequence reads obtained in next generation sequencing. Dohm et al. (2008) considered two Solexa read data sets and found that error rates were greater at the end of reads (0.3% at the beginning compared with 3.8% at the end) and wrong base calls are often preceded by base G. Also, base substitution errors were significantly disproportionate with A to C substitution error being 10 times more frequent than the C to G substitution. Similar artifacts were observed by Bravo and Irizarry (2009) who considered data from the control lane of an Illumina ChIP-seq experiment and reported A to T miscall to be the most common error in their calibration study. They also reported that the error rates vary with the position on the read and questioned the utility of the quality scores supplied by the manufacturers with a base call. These and other systematic biases may lead to wrong statistical conclusions. Finally, Oshlack and Wakefield (2009) considered three data sets including sequencingdata from Illumina and SOLiD and demonstrated for each data set that when gene expression is calculated using aggregated tag counts for each gene in RNA-seq technology the ability to call differentially expressed genes (or ranking) between samples is strongly associated with the length of the transcript.
Statistical tools for using sequence reads
There are a number of notable papers in the area of transcriptome analysis using NGS technology: Nagalakshmi et al. (2008) in yeast; Cloonan et al. (2008); Morin et al. (2008); Marioni et al. (2008) in human; Mortazavi et al. (2008) in mouse; Vera et al. (2008) in butterfly, and so on. A next generation sequencing technology obtains millions of short reads from the transcript population of interest and by mapping these reads to the genome, RNA-Seq produces digital (counts) rather than analog signals and offers the chance to detect novel transcripts. Obviously, there are several protocols for transcript quantification for NGS data
Mapping software such as MAQ by Li et al. (2008) are useful in assembling short sequence reads to match a reference genome. MAQ uses a Bayesian calculation to produce a phred-scaled probability (–10 times the common logarithm of the probability) that an individual alignment is mapped incorrectly. It also includes the capability to use mate-pair information for paired-end read alignment in diploid samples. MAQ can assimilate the mapping quality and raw sequence base quality scores and uses a Bayesian analysis to make a final genotype call. More recent mapping tools such as Bowtie (Langmead et al., 2009) and BWA (Li and Durbin, 2009) utilize computational advantages of string matching theory via the Burrows-Wheeler transform to provide much faster algorithms for short read alignment against a large reference genome with a small memory footprint. Bowtie extends the Burrows-Wheeler transform for alignment using backtracking to allow mismatches as well as double indexing to avoid excessive backtracking. Although it is a greedy algorithm which will not necessarily find the best match if it is inexact, Bowtie gives users options to improve its accuracy in return for computational costs. The BWA algorithm uses an efficient backward search with the Burrows-Wheeler transform and allows for inexact matching and gapped alignment. Li and Durbin (2009) describe the computational performance and accuracy of MAQ, Bowtie, and BWA on some simulated and real data examples.
The software package F-seq, developed by Boyle et al. (2008), employs kernel smoothing in converting high-throughput sequencing reads into continuous signals along a chromosome whose output can be displayed directly in the UCSC Genome Browser. This type of data summary will be useful to identify specific sequence features, such as transcription factor binding sites (ChIP-seq) or regions of open chromatin (DNase-seq). F-seq provides a more statistically rigorous tool to researchers who would otherwise use histograms to calculate regions of highly dense sequence reads. Also, Zhang et al. (2008a) developed MACS (Model-based Analysis of ChIP-seq) that utilizes Poisson modeling and to capture local biases in the genome resulting in more robust predictions of binding sites.
Jiang and Wong (2009) used statistical inference of isoform expression using high throughput RNA sequencing (RNA-Seq) data by Poisson modeling and solving a convex optimization problem. The measure RPKM (reads per kilobase of the transcript per million mapped reads to the transcriptome) was originally introduced by Mortazavi et al. (2008). Normalizing the counts of reads mapped to a gene (or to exons belonging to gene) against the transcript lengthand the sequencing depth, this RPKM measure can compare the expression measures across different genes and different experiments. However, reads that are mapped to a gene are frequently shared by multiple isoforms. Consequently, Jiang and Wong (2009) developed the following statistical model for this isoform expression estimation problem. Let G be the set of genes and F be the set of isoforms for all possible isoforms for all genes. Let lf be the length and let kf be the number of copies of the transcripts in the form of an isoform f ε F. Assuming every read is independently and uniformly sampled from all possible nucleotides in the sample, the probability that a read comes from isoform f is kf lf / L, where the total length of the transcripts in the sample is L = ΣfεF k f lf . If w denotes the total number of mapped reads, then the number of reads coming from a region of length l in f can be modeled by a Binomial random variable with w trials and probability of success kf l / L. Furthermore, if w is large and p is small, the law of rare events allows this distribution to be approximated by a Poisson distribution with mean ε =kf lw /L. Now, assume that there are m exons with respective lengths l1,...,lm and n isoforms with respective expressions θ1,...,θn. The set of observations falling into a region can be modeled by a Poisson random variable with mean
λ= lgw∑n f=1 cfgθ f where cfg is an indicator variable that equals 1 if isoform f contains exon g and equals 0 otherwise. The counts for exon-exon junctions can be modeled by a Poisson random variable with mean λ= lw∑nf=1cfgcfhθ f. In the multiple isoform case, numerical methods (e.g., hill climbing) must be used to obtain the maximum likelihood estimate of the θ’s; fortunately, the joint log-likelihood is concave, so any local maximum is also guaranteed to be a global maximum. Standard numerical calculations based on the Fisher information matrix can be problematic when some of the isoforms have low expressions, so in these cases, a Bayesian alternative using importance sampling is proposed for making statistical inferences.
In a recent article, Bullard et al. (2010) explore the effects of different systematic sources of variability in measuring the differential expression of genes using three platforms (mRNA-seq data from Illumina sequencing, microarray and quantitative real time PCR assay data), all of which are based on the context of the Microarray quality control project (MAQC). In addition, it is also shown that using an auto-calibration instead of Illumina’s standard way of reserving one flowcell lane for the control can help improving the mapping quality of the reads thereby ensuring a much more cost-effective and efficient experimental design. Normalization strategies are suggested to get rid of these biases
Other notable contributions leading to broad data analytic tools include Johnson et al. (2007, mapping techniques); Fejes et al. (2008, enrichment analysis); Ji et al. (2008, ChIP-seq data); Sharon et al. (2008, protein-DNA interactions); Zhang et al. (2008b, ChIP-seq data); Rozowsky et al. (2009, ChIP-seq data); Langmead et al. (2009, alignment tool); Xie and Tammi (2009, DNA copy number variation). For a comprehensive review of methods for ChIP-seq and RNA-seq data, see Pepke et al. (2009).
R and bioconductor packages
Already, a number of R (http://www.r-project.org/) and Bioconductor (http://www.bioconductor.org/) packages/tools for analyzing NGS data have been developed. The rtracklayer (Lawrence et al., 2009) package provides an interface between R and genome browsers. This package includes functions that import/export, track data and control/query external genome browser sessions/views. The chipseq (Kharchenko et al., 2008) package useful tools for design and analysis of ChIP-seq experiments and detection of proteinbinding positions with high accuracy. These tools include functions that improve tag alignment and correct for background signals. The Biostrings 2 (Pages, 2009) package allows users to manipulate big strings easily and quickly by introducing new implementations and new interfaces into Biostrings 1. The ShortRead package (Morgan et al., 2009) provides useful tools for analyzing highthroughput data produced by Solexa, Roche 454, and other sequencing technologies. These tools include input and output, quality assessment, and downstream analysis functions. The IRanges package (Pages et al., 2009) includes functions for representation, manipulation, and analysis of large sequences and subsequences of data as well as tools for attaching information to subsequences and segments. The BSgenome package (Pages, 2009) provides infrastructure for accessing, analyzing, creating, or modifying data packages containing full genome sequences of a given organism. The biomaRt package (Durinck et al., 2006) allows users to connect to and search BioMart databases and integrates them with software in Bioconductor. This package includes functions that annotate identifiers with genetic information and allow retrieval of data on genome sequences and single nucleotide polymorphisms. The ChIPpeakAnno package (Zhu et al., 2009) provides users with facilitation tools for the batch annotation of the peaks identified from either ChIP-chip or ChIP-seq experiments. These tools include functions that find the nearest gene, exon, miRNA or transcription factor binding sites as well as identify Gene Ontology (GO) terms followed by GO enrichment test. The TileQC package (Dolan and Denver, 2009) can be used with Solexa output; it identifies bias and error in data by flow cell tiles through graphical means. The PICS package (Zhang et al., 2010; http://www.bioconductor.org/packages/2.6/bioc/html/PICS.html) can identify enriched regions by extracting information from ChIP-Seq aligned-read data via a Bayesian hierarchical t-mixture model. The rGADEM package (Droit et al., http://bioconductor.org/packages/2.6/bioc/html/rGADEM.html) provides users with an efficient de novo motif discovery tool for large-scale genomic sequence data. Several of these packages work in consort as shown in Figure 4.

Figure 3: Joint modeling of ChIP-seq and ChIP-chip data.

Figure 4: The dependency among the released R/Bioconductor packages. The solid lines represent the direct dependency and the dotted lines the indirect dependency.
Besides, there are several packages/tools for visualizing NGS data. The HilbertVis package (Anders, 2009) provides several functions for visualizing long vectors of integer data by means of Hilbert curves. The GenomeGraphs packages (Durinck et al., 2009) allows users to plot different data types such as array CGH, gene expression, sequencing and other data, together in one plot using the same genome coordinate system. These tools include functions to convert the Eland and Q-score data contained within the Solexa text files to a more flexible database form.
Some selected applications and statistical analyses
In earlier sections, we have mentioned several papers (and packages) developing statistical tools for use with NGS data, many of which are broad based while others are specific to certain types of applications. In this section, we selectively review a number of additional papers applying the next generation sequencing technology in a multitude of biological investigations along with brief descriptions of the statistical techniques used; each of these papers employ interesting novel statistical methods for downstream analyses of NGS data for solving the problem at hand. For a general review of applications of NGS technology, see the article by Fox et al. (2009).
In a recent article, Choi et al. (2009) used NG ChIP-seq data together with array hybridization data towards enhancing the detection of transcription factor binding sites. There are a number ofreasons for combining these two platforms. ChIP-seq offers genomewide coverage in a single base pair resolution at low cost; however, with ChIPseq, different mapping strategies may identify mutually exclusive peak regions as candidate binding sites and massively parallel sequencing may not work well for all DNA fragments uniformly. Other mapping methods not relying on direct sequencing, e.g. ChIP-chip, can be a valuable source to complement the weakness of the sequencing technology. See Schones and Zhao (2008) for an excellent review of various technologies and their combination for studying chromatin modifications genome-wide. This rather interesting analysis by Choi et al. (2009) uses a hierarchical hidden Markov model to combine individual hidden Markov models used with each data types. Regular hidden Markov models (HMMs) have been a standard tool in modeling ChIP-chip data (Humburg et al, 2008). The main difficulties in combining data from these two sources arise from the distinct nature of these two data types. The peaks identified by ChIP-seq are expected to form regions that are much sharper than those in ChIPchip due to its superior resolution, whereas ChIP-chip tends to report broader regions with moderate significance including potential false positives. The signals from the two data sources have to be appropriately weighted in order to keep the overall false positive rates low and obtain good sensitivity in the joint analysis. This is done through a mostly Bayesian strategy. Individual HMMs {hst} and {hct} are fit to both ChIP-seq data {St} and ChIP-chip data {Ct} which, in turn, are controlled by a master or hierarchical HMM {ht} consisting of either ChIP enriched or background states (Figure 3). The states in the individual HMMs were generated from a multinomial distribution given the emissions of the master HMM. HMM in ChIPchip followed the uniform and normal distributions, respectively, for the ChIP enriched and the background states. The counts in ChIPseq data in the two states were modeled by a generalized Poisson and a zero-inflated Poisson (to reflect the empty reads), respectively. Posterior probabilities of the master states are computed and a state is declared to be ChIP enriched if this probability exceeds a given threshold, say, 90%.
A similar combination of data types was used by Zang et al. (2009), who looked for spatial clusters of signals, for identification of ChIP enriched signals for histone modification profiles. Chu et al. (2009) applied whole genome sequencing to diagnose the fetal genetic disease using cell-free DNA from maternal plasma samples in the first trimester of pregnancy. Cokus et al. (2008) used NG sequencing to identify novel components of the Arabidopsis for methylation. In a rather potentially high impact application, Quon and Morris (2009) developed a statistical method to identify the primary origin of a cancer sample via next generation sequencing. This utilizes a detail profile of tissues of each primary origin and not a data based classifier. Friedländer et al. (2008) used deep sequencing technology to identify small RNAs (miRNAs). They were able to identify and experimentally validate four novel miRNAs for the worm Caenorhabditis elegans and altogether over two hundred potential miRNAs using data from C. elegans, dog and human those were previously unknown. They computed a test statistic (i.e., a score) based on the compatibility of the position and frequency of sequenced RNA with the secondary structure of the miRNA precursor. The false positive rates (or sizes) of their test were estimated using a permutation algorithm. Meng et al. (2008) studied targeted gene inactivation in zebra fish using engineered zinc-finger nucleases (ZFNs). They demonstrated that coinjection of mRNAs encoding these ZFNs (that were engineered to recognize certain sequence) into one-cell-stage zebra fish embryos led to mutagenic lesions at the target site that were transmitted through the germ line with high frequency. They showed this by comparing the Solexa sequence data from target sites versus off-target sites at each ZFN dose; Fisher’s exact test was employed to test whether these two groups had different insertion/deletion rates in the sequence. Dalevi et al. (2008) considered the problem of matching individual short reads sampled from the collective genome of a microbial community to protein families. They found that assignments based on proxygenes, where full-length protein sequences with high similarity to the translated sequences are identified, were typically more accurate than direct assignment. However, proxy-gene assignments may lead to redundancy, so hierarchical clustering was used to significantly reduce the size of the dataset while still maintaining the quality of the functional information obtained from the analysis. Use of NGS for discovering structural variation is reviewed in Medvedev et al. (2009). Very recently, Goya et al. (2010) developed novel statistical methods of predicting single nucleotide variant from NGS data using mixtures of binomial distributions to model allelic counts. Their methodologies were developed specifically to work with cancer data where earlier simpler methods (e.g., Li et al., 2008) did not work adequately.
With new technology come new challenges for the data analysts and next generation sequencing is no exception. There seems to be a general perception that given the high quality of NGS data, replication is hardly necessary. While this may be true for technical replicates it cannot be the case with biological replicates in experiments where a conclusion is being reached about certain genetic aspect of a population from a biological sample from that population. The high dimensionality of the data makes direct use of classical statistical techniques difficult if not outright impossible. The success stories thus far seem to come from mostly Bayesian statistical techniques; however, often these are combined with frequentistcalculations. In many instances, the entire analysis combines various statistical methods of varied complexities in a mostly ad hoc manner. Although, simulation studies are generally performed to demonstrate the effectiveness of the combined approach, its overall statistical properties are difficult to assess from a theoretical standpoint; in particular, no assessment of optimality of the overall statistical procedure can be assessed this way.
There is also a misconception amongst some practitioners that Bayesian methods are immune from the sample size requirement. While it is true that one can always get a Bayesian answer even with a small number of biological replicates, for good empirical statistical properties such as posterior consistency, a large sample size is necessary; this issue is directly linked with overall robustness with respect to prior misspecification and the overall reliability of the answers from a Bayesian calculation. Next generation sequencing also presents some of the same statistical challenges presented by other high throughput genomic data types, namely, high dimensionality, global error rate control, and correlation amongst counts at different sites.
The challenges described above also present new opportunity for the statisticians for collaborative (interdisciplinary) as well as methodological development in this exciting area of research. In particular, there is a need for development of systematic statistical methods that adhere to fundamental statistical principles while addressing the practical needs of the researchers. There is still scope of employing novel statistical methods as new applications to this technology emerge. Also, methods to get a better handle of the issues mentioned in the previous paragraph are needed sooner than latter. Finally, there needs to be more work towards development of study designs and establishing global statistical standard in these platforms.
We acknowledge funding from National Science Foundation (grant DMS- 0805559 to Susmita Datta) and National Institute of Health (grant numbers CA133844 and 1P30ES014443 to Susmita Datta). We thank the reviewers for their constructive comments.