Advanced Techniques in Biology & Medicine
Open Access
ISSN: 2379-1764
ISSN: 2379-1764
Review Article - (2018) Volume 6, Issue 2
β1-adrenergic receptor (Adrb1), a member of the G-protein coupled receptor (GPCR) superfamily, is a critical regulator of heart function. All GPCRs are phosphorylated at multiple sites and the specific pattern of phosphorylation acts as a “barcode” to regulate receptor function and downstream physiological processes in a tissue-specific manner. However, little is known about the location and function of Adrb1 phosphorylation sites in vivo due to the lack of specific antibodies. As a first step to identify the phosphorylation states of Adrb1 and associated functions in the in vivo mouse heart, we developed the following experimental strategy: 1) identification of agonist-dependent Adrb1 phosphorylation sites in isolated perfused mouse heart using advanced phosphoproteomics techniques; 2) definitive assignment of these phosphorylation sites by high-quality mass spectrometry (MS) data obtained from Adrb1- overexpressing HEK 293T cells; 3) generation of knock-in (KI) Mice expressing Adrb1 fused with FLAG-tag at the N-terminus for immunoaffinity purification to reveal phosphorylation status within the living organism; 4) elucidation of phosphorylation levels at specific sites of Adrb1 in KI mouse heart by MS measures of phosphorylated peptide to corresponding unphosphorylated peptide ion intensity ratios. Using this strategy, we identified Ser462 at the C-terminus of Adrb1 as an agonist-dependent phosphorylation site in the perfused mouse heart. We also revealed the basal phosphorylation ratios at Ser274 (0.25), Ser417 (0.55) and Ser462 (0.0023) in KI Mice. These findings provide novel insights into the regulatory mechanisms of Adrb1 function mediated by site-specific phosphorylation.
Keywords: β1-Adrenergic receptor; G protein coupled receptor; Phosphorylation; Proteomic technology; Knock-in mice; Immunoaffinity purification
Adrb1: β1-Adrenergic Receptor; ADRB2: Beta- 2 Adrenergic Receptor; GPCR: G-Protein Coupled Receptor; HEK 293T: Human Embryonic Kidney 293T; HPLC: High Performance Liquid Chromatography; ISO: Isoproterenol; KI: Knock-In; MS: Mass Spectrometry; Pln: Phospholamban; PTM: Post-Translational Modification; Ryr2: Ryanodine Receptor 2; SILAC: Stable Isotope Labeling by Amino Acids in Cell Culture
Previous studies have mapped the phosphorylation sites and associated site-specific functions of the beta-2 adrenergic receptor [1,2]; however, these phosphorylation sites are less well characterized in ADRB1. Serine 312 in the third intracellular loop of human ADRB1 is the only residue reported to be a phosphorylation substrate of protein kinase A. While Ser312 has been shown to regulate recycling and functional re-sensitization in an overexpression system [3], the identity and physiological relevance of other sites remain uncertain.
Specific patterns of phosphorylation determine the intracellular signaling-mediated events initiated by seven-trans membrane G-protein coupled receptors (GPCRs) as well as receptor desensitization, Endocytosis and intracellular trafficking [4,5]. Putative GPCR phosphorylation sites predicted based on sequence motifs have been validated mainly by site-directed mutational analysis in cell lines [6].
In vitro reconstitution has proven to be a powerful tool for revealing phosphorylation sites and potential functions; however, this in vitro strategy cannot definitively identify all residues phosphorylated in living organs [7] as GPCRs are heterogeneously phosphorylated at multiple sites for flexible tissue-specific regulation, termed the “phospho-barcode” hypothesis [1,8]. Despite in vitro experiments supporting the phospho-barcode, there may be significant differences in phospho-regulation between in vitro and in vivo environments, so additional in vivo studies are required to elucidate the phosphorylation patterns of receptors and associated physiological responses. These differences may underlie one ubiquitous challenge that has plagued drug development, the discrepancies in drug efficacy between in vitro and in vivo conditions. Although direct analysis of in vivo phosphorylation state is the ideal, it is exceedingly difficult due to the relatively low levels of most phosphorylated proteins compared to their unphosphorylated counterparts under physiological conditions [9,10]. In addition, functional analysis in tissues is challenging owing to the complexity and wide dynamic range of phosphorylated peptides.
Mass spectrometry (MS) and selective enrichment of phosphorylated proteins are powerful tools to address these challenges. Recent advances in MS have made it possible to analyze signaling pathways by facilitating high-throughput identification of phosphorylation sites with high accuracy and sensitivity [11,12]. Furthermore, the revolutionary development of post-translational modification (PTM) enrichment methods, such as immobilized metal affinity chromatography and immunoprecipitation using phospho-specific antibodies [13], has enabled the identification and quantification of many PTMs.
Although putative phosphorylation sites of ADRB1 have been identified using site-directed mutagenesis in cell lines [3,14], there have been no reports on Adrb1 phosphorylation sites in mammalian tissues. To overcome these limitations, we have developed an experimental strategy to identify Adrb1 phosphorylation sites in isolated perfused mouse heart utilizing advanced phosphoproteomics techniques [15].
Antibodies are powerful tools for evaluating protein expression levels and phosphorylation status in vivo. However, attempts at identifying these sites on Adrb1 have failed using commercially available antibodies, possibly due to insufficient specificity or low expression level of the receptor in vivo. To overcome these difficulties, we have generated KI Mice expressing Adrb1 fused with FLAG-tag at the N-terminus (FLAG-Adrb1 KI Mice). In a KI model, the target gene is inserted via homologous recombination, resulting in a natural expression pattern and level [16], while the FLAG epitope fused inframe with Adrb1 enables anti-FLAG immunoaffinity purification to facilitate identification of PTMs in vivo. We estimated the site-specific phosphorylation levels of Adrb1 by measuring the ion intensity ratios of phosphorylated peptides relative to corresponding unphosphorylated counterparts using MS. Although the ratio does not exactly translate into stoichiometry [17], it does provide a relative measure of high- and low-phosphorylation sites under specific conditions such as agonist stimulation. In this review, we demonstrate the efficacy of this strategy for determining the phosphorylation states of Adrb1 under basal and agonist-stimulated conditions.
Identification of Phosphorylation Sites on Adrb1 in Mouse Heart
As a first step to identify Adrb1 phosphorylation sites in vivo under physiological conditions, we established a Langendorff heart perfusion system and compared the phosphorylation states of peptides isolated from cell membranes of control hearts to hearts treated with the Adrb1 agonist isoproterenol (ISO). After proteolysis of membrane proteins, the phosphorylated peptides were enriched using modified hydroxyl acid-modified metal oxide chromatography [18] followed by strong cation exchange chromatography fractionation. As the stoichiometric level of phosphorylated protein is extremely low compared to the unmodified protein, it is necessary to reduce the sample complexity for successful MS analysis. This system was further validated by measuring the phosphorylation levels of ryanodine receptor 2 (Ryr2) and phospholamban (Pln), major determinants of cardiac contractility and relaxation. Phosphorylation levels at both Ser16 and Thr17 in Pln increased significantly following ISO stimulation, consistent with the results of a previous study [19]. Isoproterenol also phosphorylated multiple sites in Ryr2 (S2807, T2809 or S2810, S2013 and S2821), consistent with a previous report demonstrating that Ryr2 is phosphorylated by calcium/calmodulin-dependent protein kinase type II, a protein activated downstream of β-adrenergic receptor stimulation [20]. A total of 11,295 phosphorylated peptides from 3,925 proteins were identified from the crude membrane protein fraction of four mouse hearts. Using this enrichment and fractionation approach, four phosphorylated peptide fragments in the C-terminus and third cytoplasmic loop of Adrb1 were identified (Table 1). However, ISO enhanced phosphorylation only at Ser274, S278 and S462. These findings suggest that Ser274, S278 and S462 are phosphorylated as a consequence of Adrb1 activation in heart.
Confirmation of Phosphorylation Sites in Adrb1- Overexpressing HEK 293T Cells
To confirm the location of these phosphorylation sites and provide additional evidence for their physiological relevance, we examined phosphorylation patterns in HEK 293T cells overexpressing Adrb1, a system that allows for the isolation of sufficient amounts of protein for high sequence coverage of tryptic peptides and high-quality MS spectra. The phosphorylation states were analyzed quantitatively in control and ISO-treated HEK 293T cells transfected with a plasmid expressing Adrb1 using stable isotope labeling by amino acids in cell culture (SILAC) labeled tryptic peptides as an internal standard prepared independently from Adrb1-overexpressing cells. Table 1 lists the phosphorylated peptides identified in both mouse heart and Adrb1-overexpressing HEK 293T cells. Seven phosphorylated sites located at the C-terminus and the third cytoplasmic loop of Adrb1 were reproducibly identified in both mouse heart and Adrb1-transfected HEK 293T cells: Ser274, Ser280, Ser412, Ser417, Ser450, Ser451 and Ser462 (Figure 1). These conserved phosphorylation sites provide important clues to Adrb1 signaling mechanisms that may aid in drug discovery. Among them, ISO induced a 2-fold increase in Ser462 phosphorylation compared to control cells. Further, this site was phosphorylated in the perfused heart system only when stimulated with ISO. These findings indicate that Ser462 in the C-terminus of Adrb1 is an agonist-dependent phosphorylation site in vivo and in vitro. Although phosphorylation levels at Ser274 and Ser280 were strongly increased in ISO-stimulated mouse heart, levels were similar in ISO-stimulated and control Adrb1- overexpressing HEK 293T cells (Table 1). We speculate that the fetal bovine serum supplementing the culture medium may have induced continuous phosphorylation at these sites. On the contrary, the basal levels of Ser274 and Ser280 phosphorylation in perfused heart were reduced completely in medium that did not contain activators. We presume that these sites may be phosphorylated by a kinase other than G protein-coupled receptor kinases that specifically recognize and phosphorylate agonist-activated GPCRs.
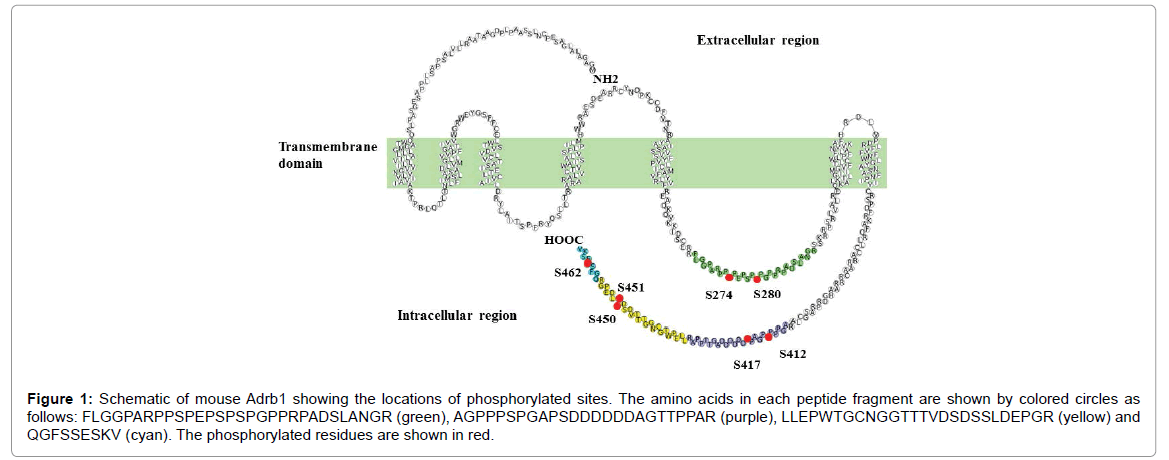
Figure 1: Schematic of mouse Adrb1 showing the locations of phosphorylated sites. The amino acids in each peptide fragment are shown by colored circles as follows: FLGGPARPPSPEPSPSPGPPRPADSLANGR (green), AGPPPSPGAPSDDDDDDAGTTPPAR (purple), LLEPWTGCNGGTTTVDSDSSLDEPGR (yellow) and QGFSSESKV (cyan). The phosphorylated residues are shown in red.
| Sequence | Position | Phosphorylation Site | m/z | Mouse Heart | HEK-293T | |||
|---|---|---|---|---|---|---|---|---|
| Charge | Intensity | Ratio ISO/Ctrl |
||||||
| Start | End | Ctrla | ISOb | |||||
| YLAITSPFR | 157 | 165 | S162 | 574.28 | 2 | 1.2 | ||
| YQSLLTR | 166 | 172 | S168 | 480.73 | 2 | 1.2 | ||
| KIDSCER | 257 | 263 | S260 | 494.2 | 2 | 1.2 | ||
| FLGGPARPPSPEPSPSPGPPRPADSLANGR | 265 | 294 | S274, S280 | 1046.83 | 3 | ND | 1.00E+07 | 0.9 |
| FLGGPARPPSPEPSPSPGPPRPADSLANGR | 265 | 294 | S274 | 765.38 | 4 | ND | 1.00E+07 | 1.2 |
| RPSRLVALR | 299 | 307 | S301 | 383.22 | 3 | 1.3 | ||
| ASGCLAR | 400 | 406 | S401 | 407.67 | 2 | 1.9 | ||
| AGPPPSPGAPSDDDDDDAGTT†PPAR | 407 | 431 | S412, S417, T426 (T427)† | 872.65 | 3 | 1.1 | ||
| AGPPPSPGAPSDDDDDDAGTTPPAR | 407 | 431 | S412, S417 | 845.99 | 3 | 2.30E+07 | 2.20E+07 | 1.1 |
| AGPPPSPGAPSDDDDDDAGTT†PPAR | 407 | 431 | S412, T426 (T427)† | 845.99 | 3 | 1.1 | ||
| AGPPPSPGAPSDDDDDDAGTT†PPAR | 407 | 431 | S412, T426 (T427)† | 845.99 | 3 | 1.1 | ||
| AGPPPSPGAPSDDDDDDAGTTPPAR | 407 | 431 | S412 | 819.34 | 3 | 0.9 | ||
| AGPPPSPGAPSDDDDDDAGTTPPAR | 407 | 431 | S417 | 819.34 | 3 | 6.30E+07 | 6.20E+07 | 1.2 |
| LLEPWTGCNGGTTTVDSDSSLDEPGR | 432 | 457 | S450 | 948.74 | 3 | 2.60E+07 | 5.70E+07 | 1.1 |
| LLEPWTGCNGGTTTVDSDSSLDEPGR | 432 | 457 | S451 | 948.74 | 3 | 1.00E+07 | 1.10E+07 | 1 |
| QGFSSESKV | 458 | 466 | S461 | 524.72 | 2 | 1.7 | ||
| QGFSSESKV | 458 | 466 | S462 | 524.72 | 2 | ND | 8.70E+06 | 2 |
aIntensity of MS chromatogram
bPeak intensity ratio of MS chromatogram
Phosphorylation sites are shown in red
ND: Not Detected; †: The phosphorylated site at the vicinal threonine could not be identified.
Table 1: List of phosphorylated Adrb1 residues in mouse heart and Adrb1-overexpressing HEK 293T cells.
On the other hand, many phosphorylation sites on Adrb1 not detected in mouse heart were identified in Adrb1-overexpressing HEK 293T cells. These sites may be phosphorylated only under unphysiologically high expression levels. Alternatively, phosphorylation levels at these sites may be too low to detect in the mouse heart. In any case, the agonist-induced phosphorylation at Ser462 in both mouse heart and Adrb1-overexpressing cells strongly suggests critical involvement in physiological signal transduction pathways, an issue that merits further investigation.
Generation of FLAG-Adrb1 KI Mice
The processes of GPCR activation, internalization and trafficking are well characterized in vitro; however, these processes have not been examined extensively in vivo. For such in vivo analyses, KI Mice may be a useful tool. Indeed, KI Mice expressing specific GPCRs fused to green fluorescent protein have been produced to visualize the distribution and internalization of native GPCRs under agonist stimulation [21- 24]. Moreover, KI Mice with biological loss-of-function mutations have been generated to evaluate the physiological contributions of various target sites [25-27].
In response to CPCR phosphorylation, β-arrestins are recruited to the membrane, leading to receptor desensitization, inactivation and internalization. In addition, activities and interactions with other molecules are regulated by phosphorylation of the GPCR, underscoring the importance of complete phosphorylation state determination in vivo. However, this is enormously difficult due to the low stoichiometry of phosphorylated proteins under physiological conditions. Enrichment of phosphorylated peptides may be effective, although information on the expression level is lost. Thus, individual quantitative analyses of global protein expression and phosphorylation levels are required for a precise interpretation of phosphorylation changes, but it is very challenging to detect an extremely low-abundance unmodified peptide fragment using global proteomic analysis without appropriate antibodies. Alternatively, isotope dilution is a widely used method for quantitative analysis of phosphorylation state by MS [28,29] and multiple-reaction monitoring is a useful method for measuring the absolute phosphorylation occupancy at a specific site in a protein of interest [30]. However, these methods require the synthesis of isotopelabeled peptides, which is time and resource intensive and in most cases, enrichment of phosphorylated peptides is still required for lowabundance proteins.
In contrast, a simple method to assess the extent of phosphorylation is by calculating the ion intensity ratios of phosphorylated tryptic peptides relative to the corresponding unphosphorylated counterparts by MS analysis since these peptide forms are closely matched for amino acid composition and high performance liquid chromatography (HPLC) retention time. It is well known that the ionization efficiency of peptides strongly depends on modification state as well as on amino acid composition. In particular, addition of a phosphate group suppresses ionization in MS. Accordingly, the ratio qualitatively reflects phosphorylation state, although it does not directly translate into stoichiometry [17]. Moreover, MS analysis of KI tissue provides information on steady-state phosphorylation, although it is less useful for rapid changes. Thus, KI Mice expressing Adrb1 with an N-terminal FLAG epitope adequate for immunoaffinity purification [31,32] were generated based on the assumption that the small FLAG-tag would not disrupt the in vivo function of Adrb1.
We inserted a construct containing the chimeric intron, signal sequence and mouse Adrb1 CDS with three repeated FLAG epitope tags into the Adrb1 locus (Figure 2). In this KI model, the promoter region and 3' UTR of Adrb1 was contained in the targeting construct, so we speculated that proper Adrb1 protein localization would be maintained. Alternatively, it was unclear whether expression levels would be influenced by transcriptional regulatory elements of the targeting construct (such as the promoter, intron and polyadenylation signal sequence) [33]. Indeed, the generated KI Mice showed elevated Adrb1 mRNA compared to wild types, so protein expression, intracellular distributionand function of Adrb1 in KI Mice require further investigation. However, membrane localization appeared normal as discussed below.
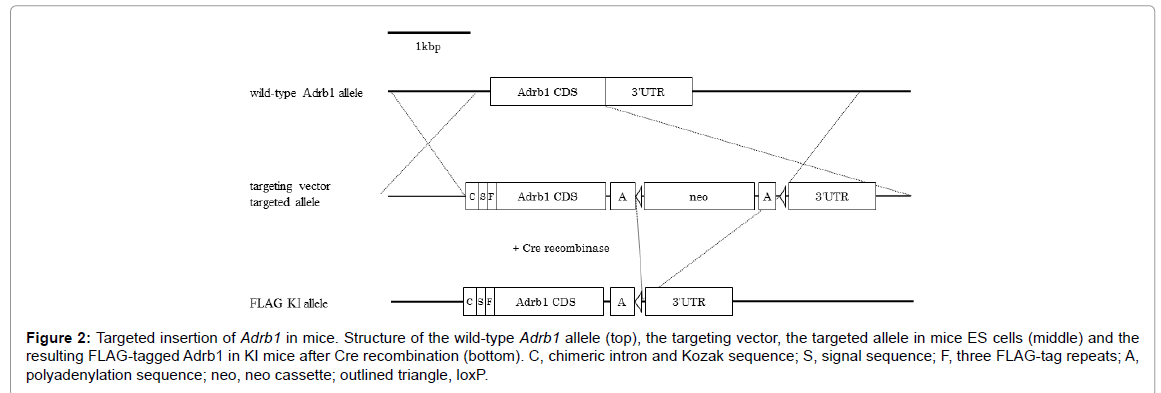
Figure 2: Targeted insertion of Adrb1 in mice. Structure of the wild-type Adrb1 allele (top), the targeting vector, the targeted allele in mice ES cells (middle) and the resulting FLAG-tagged Adrb1 in KI mice after Cre recombination (bottom). C, chimeric intron and Kozak sequence; S, signal sequence; F, three FLAG-tag repeats; A, polyadenylation sequence; neo, neo cassette; outlined triangle, loxP.
Phosphorylation Levels at Specific Sites of Adrb1 in KI Mouse Heart
The phosphorylation sites on Adrb1 in KI mouse heart were identified by combining immunoaffinity purification with the high sensitivity of MS analysis. Six tryptic peptide fragments were identified and three were detected together with their phosphorylated peptides from the membrane protein fraction of KI mouse heart (Table 2 and Figure 3). All phosphorylated fragments identified in the FLAG-Adrb1 KI mouse matched those identified in isolated perfused mouse heart by TiO2 purification [15]. On the other hand, peptide fragments of Adrb1 could not be detected from the soluble protein fraction, indicating that FLAG-Adrb1 is properly localized in KI mouse heart.
| Sequence | Start | End | Phospho-site | m/z | Charge | Phosphorylation ratioa (Phospho/Non-phospho) |
|---|---|---|---|---|---|---|
| FLGGPARPPSPEPSPSPGPPRPADSLANGR | 265 | 294 | S274 | 765.38 | 4 | 0.25 ± 0.058 |
| AGPPPSPGAPSDDDDDDAGTTPPAR | 407 | 431 | S417 | 819.34 | 3 | 0.56 ± 0.070 |
| QGFSSESKV | 458 | 466 | S462 | 524.72 | 2 | 0.0023b |
| YQSLLTR | 166 | 172 | - | 440.75 | 2 | - |
| LLCCAR | 379 | 384 | - | 396.7 | 2 | - |
| LLEPWTGCNGGTTTVDSDSSLDEPGR | 432 | 457 | - | 922.09 | 3 | - |
Table 2: Tryptic peptide fragments and phosphorylation levels of FLAG-Adrb1 in KI mouse heart.
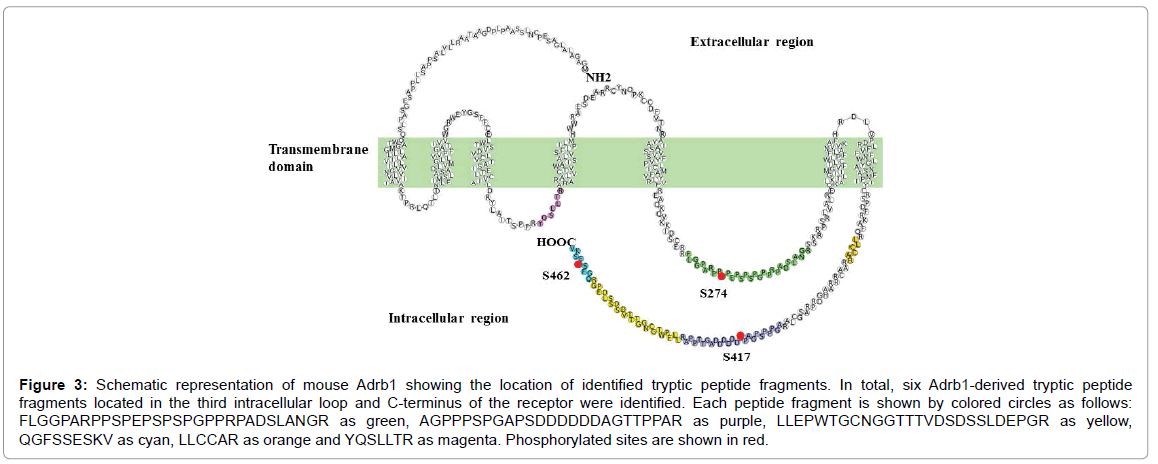
Figure 3: Schematic representation of mouse Adrb1 showing the location of identified tryptic peptide fragments. In total, six Adrb1-derived tryptic peptide fragments located in the third intracellular loop and C-terminus of the receptor were identified. Each peptide fragment is shown by colored circles as follows: FLGGPARPPSPEPSPSPGPPRPADSLANGR as green, AGPPPSPGAPSDDDDDDAGTTPPAR as purple, LLEPWTGCNGGTTTVDSDSSLDEPGR as yellow, QGFSSESKV as cyan, LLCCAR as orange and YQSLLTR as magenta. Phosphorylated sites are shown in red.
To evaluate the phosphorylation level at each site, peak intensity ratios of phosphorylated peptides to unphosphorylated counterparts were calculated (Table 2). Phosphorylation at Ser274, which was not detected in the perfused mouse heart without agonist stimulation [15], was observed at basal levels in the Adrb1 KI model. Alternatively, S462 phosphorylation level was extremely low as expected in the absence of agonist stimulation and phosphorylation at Ser450 and Ser451 was not detected in the KI mouse model. Thus, we believe that the Adrb1 phosphorylation status of KI mouse heart reflects the natural physiological phosphorylation state.
These results demonstrate that FLAG-Adrb1 KI Mice are a valuable tool for analyzing the stoichiometry of Adrb1 phosphorylation in vivo without the need for expensive radioligands.
In summary, we have identified endogenous Adrb1 phosphorylation sites in mouse heart using advanced phosphoproteomics techniques and estimated the phosphorylation levels at specific sites in vivo by generating FLAG-Adrb1 KI Mice. This mouse line could be a valuable tool for investigating the downstream signaling mechanisms and physiological functions of Adrb1 in vivo as well as receptor distribution and trafficking. The techniques described here could also be used to analyze the endogenous phosphorylation states of other GPCRs.
We would like to thank S. Okubo, S. Fujiwara, T. Hara and S. Kuboi (Takeda Pharmaceutical Company Limited), for their expert technical assistance. We also thank Dr. T. Andou (Axcelead Drug Discovery Partners, Inc.), for their encouragement and helpful discussion.