Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2016) Volume 9, Issue 7
Malaria is the one most important parasitic disease of humans, which affects approximately one hundred countries and threatens half of the world’s population. The Plasmodium aspartic protease called plasmepsins performs a vital role in providing nutrients to the malaria parasite, which make these proteins as an excellent drug target. In this study, we have carried out a comparative protein modeling, active site analysis and structural analysis of all ten plasmepsins from Plasmodium falciparum. In this report we have analyzed in-silico structure modeling and made efforts to characterize plasmepsins structure and further propose its functional information. The phylogenetic analysis and disulfide linkages indicate, plasmepsin I to IV and HAP have similar structure, function property. Whereas, plasmepsin IX to X and plasmepsin VI to VIII belong to a separate cluster. The integral membrane protein plasmepsin V has a functional characterization as compared to the others aspartic proteases from Plasmodium falciparum. The overall study summarizes the need of good model to understand the structure and function activity and to design potent small molecule inhibitors targeting all ten plasmepsins, specifically Plasmepsin V as important target.
Keywords: Plasmepsins, Plasmodium falciparum, In-silico analysis, Homology modeling
Malaria is a life-threatening disease caused by Plasmodium parasites transmitted to humans through the infected Anopheles mosquitoes, specifically in the region of tropical and subtropical climate and is active during the dusk and dawn [1]. About 20 different Anopheles species are globally important around the world [2]. Transmission is more intense in places where the mosquito lifespan is longer, because the parasite has time to complete its development inside the mosquito and it prefers to bite humans rather than other animals [3,4]. Resistance to antimalarial medicines is a recurring problem. In recent years, parasite resistance to artemisinins has been detected in 5 countries. If resistance to artemisinins develops and spreads to other large geographical areas, the public health consequences could be dire [5,6]. World Health Organization (WHO) recommends the routine monitoring of antimalarial drug resistance and supports countries to strengthen their efforts in this important area of work [7]. Effective drug is a critical component of malaria control. Selection of Plasmodium sp. parasites resistant to multiple drugs calls for accelerated efforts to develop new anti-malarial drugs targeting novel essential parasite pathways [8]. In humans, the disease is the result of infection by Plasmodium falciparum (Pf), Plasmodium malariae, Plasmodium ovalae or Plasmodium vivax. Plasmodium knowlesi, majorly known as the fifth human malaria parasite. Of these species, Plasmodium falciparum is the most lethal and considered as important target for drug intervention [9-13].
During the intra-erythrocytic stage of infection, the malaria parasite Plasmodium falciparum digests most of the host cell hemoglobin. Hemoglobin (Hb) degradation is essential for the growth of the malarial parasites. The degradation process that occurs inside an acidic digestive vacuole is thought to involve the action of aspartic proteases of Plasmodium, termed plasmepsins (PMs) [14-16]. The plasmepsins perform a crucial role in the provision of nutrients for the red cell stages of the malaria parasite and thus make excellent drug targets [17]. Inhibition of aspartic proteases aid to kill parasites in human red blood cells in culture. Proteases are known to play an important role in numerous pathways and represent potent drug targets for several chronic infectious diseases. Hence, aspartic proteases are considered one among the important drug-target.
The P. falciparum genome comprises a group of 10 aspartyl proteases called plasmepsin, initially discovered by identifying the hemoglobin digestion pathway in malarial victims and have been strongly considered as potential anti-malaria drug targets. The plasmepsins I, II, IV and HAP are expressed in the erythrocytic stages of the life cycle of P. falciparum and are localized in the food vacuole [18]. Plasmepsin V is an integral membrane protein present in the endoplasmic reticulum of the parasite suggesting a role in protein processing within the parasite [19-21]. Plasmepsin V, IX and X were expressed concurrently with plasmepsin I to IV but are not transported to the food digestive vacuole. The remaining plasmepsins VI- VIII are expressed during the exo-erythrocytic cycle and their functions are unknown [22,23]. For several years, the structure-based drug design of antimalarial compounds targeting P. falciparum and the plasmepsin inhibitors have received much attention due to their potential therapeutic use [24,25]. However, our current study shows a wide range of In-silico modeling and the structure analysis of all ten plasmepsins. This may provide a good foundation for designing potential anti-malarial drugs targeting plasmepsins.
Also, in order to make the strongest and most effective drug for therapy of malaria infection it will be necessary to optimize the binding of compounds to the most critical enzyme in the parasite. Based on our study we have shown that plasmepsin V is a key enzyme in the parasite and its inhibitors will provide basic foundation for further development of malarial drug based on plasmepsin V.
Sequence retrieval and alignment
The sequence of plasmepsin from P. falciparum was obtained from the protein sequence database of NCBI (GenBank Id: AAB41811). The genome ID and genome localization of each plasmepsin was retrieved from PlasmoDB [26]. PlasmoDB is a functional genomic database for malaria parasites. The multiple alignments were carried out using CLUSTAL X [27]. The identical and similar amino acids are shaded or colored.
Fold-recognition and domain analysis
The domain composition of plasmepsins was analyzed using the SMART-Simple Modular Architecture Research Tool [28] in combination with the P. falciparum (Pfam) database [29]. The classification of plasmepsins was done by using PRED-CLASS server [30].
In-silico physico chemical characterization
For physico-chemical characterization, theoretical isoelectric point (pI), molecular weight, extinction coefficient and instability index [31] were computed using the Expasy’s ProtParam server [32].
Functional characterization
The eukaryotic and viral aspartyl protease active site was predicted using PROSITE [33,34]. The SOSUI server was employed to identify the nature and function of the protein. The transmembrane region of the plasmepsin was predicted using HMM-TM: Prediction of Transmembrane Alpha-Helical Proteins. NetNGlyc 1.0 Server predicted N-glycosylation sites using artificial neural networks [35,36]. The intrinsic protein disorder regions was predicted by FoldIndex© [37] and GLOBPLOT 2 [38]. FoldIndex is a graphic web server that discriminates between folded and intrinsically unfolded proteins which defines the mean net charge, |
Phylogenetic analysis
The sequences were aligned by Clustal X version 2.0 with default options and phylogenetic tree was constructed based on the bootstrap neighbor-joining method [43] using Molecular Evolutionary Genetics Analysis (MEGA) software version 4.1 [44]. The stability of internal nodes was assessed by bootstrap analysis with 10,000 replicates.
Homology modeling, refinement and validation
The three dimensional (3D) structure of plasmepsin V was performed based on the homology modeling in PHYRE2 Protein Fold Recognition Server [45]. The Swiss PDB viewer and PyMOL were used to visualize and refine the models. The quality and validation of the obtained model was performed using PROCHECK [46], ERRAT [47] and PROVE software’s [48,49] from “SAVES: Meta Server Structure Analysis” under NIH MBI Laboratory Server. The model was also analyzed in SuperPose [50] and DALI server [51]. The obtained 3D-model was stereo-chemically evaluated on RAMPAGE server [52], which provides a score based on proline and glycine preferential positions according to a Ramachandran plot. The Prosa Web server was used to predict the Z-score of the modeled structure [53].
Docking studies using AutoDock/Vina
Molecular docking protocols are widely used for predicting the binding affinities of ligands. The PDBQT files for protein and ligand preparation and grid box generation were done using Graphical User Interface program AutoDock Tools (ADT). ADT assigned polar hydrogens, united atom Kollman charges, solvation parameters and fragmental volumes to the protein. The prepared files were saved in PDBQT format. AutoGrid was used for the preparation of the grid map using a grid box and the grid size was set to 28 × 32 × 24 xyz points and grid center was designated at dimensions (x, y, and z): 4.055, 45.931.554 and 18.066. A scoring grid is calculated from the ligand structure to minimize the computation time. AutoDock/Vina was employed for docking using protein and ligand information along with grid box properties in the configuration file and in docking both the protein and ligands are considered as rigid. The results less than 1.0 Å in positional root-mean-square deviation (RMSD) was clustered together and represented by the result with the most favorable free energy of binding. The pose with lowest energy of binding or binding affinity was extracted and aligned with receptor structure for further analysis.
The ten plasmepsin sequences from P. falciparum were retrieved in FASTA format from the NCBI database and were analyzed using bioinformatics. The genome localization, gene ID, physiochemical features of each plasmepsin are represented in Table 1. In P. falciparum 3D7, the plasmepsin I-IV, VIII and IX genes are located in chromosome 14 whereas the plasmepsins V, VI, VII and X are localized on chromosome 13, 3, 10 and 8 respectively [54]. The protein sequence for plasmepsins vary in their length, which runs from 380 aa to 630 aa with variable molecular weight. The Isoelectric point or isoionic point (pI) is the pH at which the net charge on a protein is zero or neutral and hence, it does not show any mobility in an electric field. The computed pI of Plasmepsin I, II, IV and X (pI<7) showed acidic nature whereas the other plasmepsins (V, VI, VII, VIII, IX and HAP) having pI>7 had a basic nature. It is an important feature for any protein to know for experimental aspect of molecular biology, especially in 2D gel electrophoresis, isoelectric focusing etc. The high extinction coefficient of plasmepsin V indicates presence of high concentration of Cys, Trp and Tyr present in its sequence. The computed extinction coefficients help in the quantitative study of protein-protein and protein-ligand interactions in solution. The instability index provides an estimate of the stability of the protein and the protein whose instability index is smaller than 40 is predicted as stable [55,56]. The instability index of plasmepsin VI, IX and X was computed by the server to be above 40 which predicts that the protein may be unstable.
| Name of protein | Gene ID | Chromosome number | Genomic Localization | Protein Accession number | Aminoacid length | Molecular weight | Isoelectric Point | Ext. Coefficient | Instability index |
|---|---|---|---|---|---|---|---|---|---|
| Plasmepsin I | PF3D7_1407900 | 14 | Pf3D7_14_v3: 288,297 - 289,655 (+) | P39898.2 | 452 | 51260.9 | 6.72 | 55030 | 28.47 |
| Plasmepsin II | PF3D7_1408000 | 14 | Pf3D7_14_v3: 293,471 - 294,832 (+) | P46925.1 | 453 | 51489.7 | 5.42 | 55155 | 37.4 |
| HAP | PF3D7_1408100 | 14 | Pf3D7_14_v3: 297,468 - 298,823 (+) | CAB40630.1 | 451 | 51693.2 | 8.04 | 55030 | 37.62 |
| Plasmepsin IV | PF3D7_1407800 | 14 | Pf3D7_14_v3: 283,086 - 284,435 (+) | AAW71463.1 | 448 | 50933.2 | 5.3 | 54000 | 30.06 |
| Plasmepsin V | PF3D7_1323500 | 13 | Pf3D7_13_v3: 975,403 - 977,175 (+) | AAW71468.1 | 590 | 68480.3 | 7.7 | 88225 | 37.05 |
| Plasmepsin VI | PF3D7_0311700 | 3 | Pf3D7_03_v3: 502,698 - 505,848 (+) | XP_001351190.1 | 432 | 49432.9 | 7.56 | 38320 | 43.68 |
| Plasmepsin VII | PF3D7_1033800 | 10 | Pf3D7_10_v3: 1,351,197 - 1,353,284 (+) | AAN35526.1 | 450 | 52328.1 | 8.26 | 67980 | 33.75 |
| Plasmepsin VIII | PF3D7_1465700 | 14 | Pf3D7_14_v3: 2,658,255 - 2,660,911 (-) | AAN37238.2 | 385 | 44254.2 | 9.1 | 48165 | 37.65 |
| Plasmepsin IX | PF3D7_1430200 | 14 | Pf3D7_14_v3: 1,188,349 - 1,191,466 (+) | AAN36894.1 | 627 | 74183 | 9.25 | 69860 | 41.52 |
| Plasmepsin X | PF3D7_0808200 | 8 | Pf3D7_08_v3: 416,344 - 418,065 (-) | XP_001349441.1 | 573 | 65114 | 5.35 | 47385 | 48 |
Table 1: The basic parameters of all plasmepsins from P. falciparum where gene id, chromosome number and gene localization were retrieved from PlasmoDB. Protparam database was used to find the molecular weight, isoelectric point, extinction coefficient and instability index. The protein accession number was taken from NCBI database.
Protein may have single or multiple functional regions called as domains, which perform specific biochemical functions [57]. In our study, we performed a conserved domain analysis using Pfam database and it was observed that all the plasmepsins contain Eukaryotic aspartic protease (ASP) domain. In addition to the ASP domain, plasmepsin V also contains Xylanase inhibitor N-terminal (Taxi_N) domain. The Xylanase inhibitor N-terminal domain are mostly present in plants and have a major function to create the catalytic pocket necessary for cleaving xylanase [58,59]. The plant xylanase inhibitor proteins (XIPs) that inhibit fungal xylanase activity during rice blast fungal attack and are believed to act as a defensive barrier against fungal pathogens [60,61]. The presence of Taxi-N domain along with ASP domain in plasmepsin V indicates that it has an interesting characteristic which other plasmepsins do not have and thus it acquaints plasmepsin V as an important macro-molecule for further structure and function analysis. The domain organization, Bit score and the E-value of each domain search are shown in Table 2.
| Protein | Domain | Alignments (Start-End) |
BitScore | E-value |
|---|---|---|---|---|
| Plasmepsin I |  |
138-448 | 246.9 | 2.8e-73 |
| Plasmepsin II | |
139-449 | 250.8 | 1.8e-74 |
| HAP | |
138-449 | 214.9 | 1.5e-63 |
| Plasmepsin IV | 137-445 | 249.6 | 4.3e-74 | |
| Plasmepsin V | 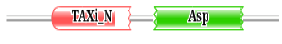 |
100-270 323-516 |
52.1 31.8 |
6.6e-14 8.3e-08 |
| Plasmepsin VI | |
99-428 | 232.4 | 7.1e-69 |
| Plasmepsin VII | |
91-443 | 169.8 | 8.1e-50 |
| Plasmepsin VIII | |
61-384 | 221.0 | 2.1e-65 |
| Plasmepsin IX | |
227-604 | 207.3 | 3.1e-61 |
| Plasmepsin X | |
247-569 | 198.6 | 1.4e-5 |
Table 2: The domain sequence alignment of plasmepsins from P. falciparum done in the pfam database where the block diagram of green color ASP represents the Aspartic protease domain and red color block Taxi_N represents the Xylanase inhibitor N-terminal domain.
The multiple sequence alignment (MSA) of plasmepsins I, II, IV and HAP has more than 60% sequence similarity whereas other plasmepsins have diversity in their sequence. In MSA analysis, the active site region residues, Asp-Thr/Ser-Gly-Ser, in all the plasmepsins were found to be conserved except in HAP (Figure 1). In case of HAP the histidine residue is present instead of aspartic acid in its active site. The SCOPE database shows that plasmepsins are pepsin like proteins which falls in pepsin_retropepsin superfamily. It is evident from the results of multiple sequence alignments that there are two-conserved motifs and some short-length conserved regions were also found. The known conserved motifs [LIVMFGAC][LIVMTADN][LIVFSA]D[ST] G[STAV][STAPDENQ][^GQ][LIVMFSTNC][^EGK] [LIVMFGTA] of aspartic protease was subjected to search against plasmepsin for motif annotation using PROSITE search. These motifs or important conserved regions might help in inference of protein structure and sequence evolution history. Based on our analysis, all the plasmepsin sequence contains two distinct active site motif detected with 10-12 amino acid length were showed in Table 3 and Figure 1. The HAP protein contains only one motif which is present in the C-terminal region of the amino acid sequence. The malaria parasite Plasmodium uses plasmepsin to degrade hemoglobin in the red blood cells. It is experimentally proven that the plasmepsins I to V and HAP are capable of cleaving native hemoglobin as well as denatured globins with an optimized pH. But the activity of other plasmepsins has not yet been demonstrated. These results indicate that all the plasmepsins have a similar active site for aspartic protease activity.
| Protein Name | Motif I | Motif II | Residue number (Start-End) |
Aspartyl proteases active site |
|---|---|---|---|---|
| Plasmepsin I | FIFDTGSANLWV | AIVDSGTSSITA | 154 – 165, 334 – 345 | 157, 337 |
| Plasmepsin II | FILDTGSANLWV | CIVDSGTSAITV | 155 – 166, 335 – 346 | 158, 338 |
| HAP | - | VILDSATSVITV | 334 – 342 | 337 |
| Plasmepsin IV | FIFDTGSANLWV | AVVDSGTSTITA | 152 – 163, 332 – 343 | 155, 335 |
| Plasmepsin V | LILDTGSSSLSF | MLVDSGSTFTHI | 115 – 126, 362 – 373 | 118, 365 |
| Plasmepsin VI | VVFDTGSSNLAI | AAIDTGSSLITG | 115 – 126, 312 – 323 | 118, 315 |
| Plasmepsin VII | VLFDTGSSQVWI | SIIDTGTYLIY | 108 – 115, 321- 331 | 111, 324 |
| Plasmepsin VIII | VLFDTGSTNLWI | AVIDTGTSSIAG | 77 – 88, 267 – 278 | 80, 270 |
| Plasmepsin IX | PIFDTGSTNIWI | LIFDSGTSFNSV | 243- 251, 492 – 503 | 246, 495 |
| Plasmepsin X | PIFDTGSTNVWV | VIFDTGTSYNTM | 263- 271, 454 – 465 | 266, 457 |
Table 3: The conserved motif pattern of each plasmepsins and the residues number of aspartic acid responsible for the protease activity.

Figure 1: The multiple sequence alignment of the representative plasmepsin sequence performed with CLUSTAL X. Active site residues are indicated by an asterisk and the conserved residues which are common to all sequences are shadowed.
The prediction of subcellular localization, disulfide linkage and glycosylation are shown in Figure 2. The online server Phobius predicted subcellular localizations which revealed that all plasmepsin I to V, IX and X are transmembrane proteins. Plasmepsin V is an integral membrane protein located in the endoplasmic reticulum of the parasite. Plasmepsin VI, VII and VIII contain no transmembrane region but they have a signal peptide region in their N-terminal. This signifies the fact that the plasmepsins which express in the erythrocytic cycle have transmembrane domain while others that express in exoerythrocytic cycle have a signal peptide instead of transmembrane domain. Only plasmepsin V is predicted to have two transmembrane spanning regions which keep this protein separate from the other plasmepsins.

Figure 2: The sub-cellular localization, glycosylation and disulfide linkages are predicted using various tools. The sub-cellular localization (Red: Signal peptide region, Blue: Non-cytoplasmic region, Magenta: Transmembrane region and Green: Cytoplasmic region). Glycan chains are shown in the glycosylated regions and the “C” letter indicates cysteine residues and cysteine disulphide bridge are connected using lines.
The disulfide bridge formation in a protein may play major role in the thermos-stability, functionality and structural stability of proteins. We calculated the cysteine residues and the disulfide bridges using various online tools like DIANNA server, DISULPHIDE and CYS_REC. The possible pairing and pattern with probability resulted in plasmepsin I- IV and VI- VIII having two disulfide bridges. Plasmepsin IX and X have four disulfide bridges were as plasmepsin V has five disulphide bridges. The disulphide architecture of plasmepsin IX is closely similar to plasmepsin V. The analysis also demonstrated that these proteins are glycosylated and most of the glycosylated proteins are known to be involved in various cellular biological functions like protein folding, cell-cell interactions, cell recognition and host-pathogen interactions.
The disorder predictors and unfoldability of protein have been proven to be useful in advancing our understanding of disordered regions with potential impact to improve the success rate of structural genomic efforts. The formation of protein crystals can be obstructed by the presence of highly flexible and disordered regions. The understanding of disordered regions as a result of structural bioinformatics efforts allowed us to extract and analyze patterns associated with these regions which can further help to overcome several potential bottlenecks for a successful structural genomics and X-ray crystallography. The unfoldability and disorder prediction of plasmepsins shows that plasmepsin IX has highly unfolded structure with the presence of six disordered regions [62]. The predicted secondary structure composition of plasmepsins was determined using the NPS@ server and Gor IV method which generates percentage of alpha helix, beta sheets and TM helix present. The results revealed that 40% beta sheets dominated among the secondary structure followed by 15–20% alpha helix. The disordered regions and the percentage of secondary structure of each plasmepsins are shown in Table 4.
| Protein | Unfoldability* | Region of disorder segment | Percentage of Alpha helix | Percentage of Beta strand | Percentage of TM helix |
|---|---|---|---|---|---|
| Plasmepsin I | 0.193 | [78]-[118], [163]-[174], [287]-[298], [300]-[309] |
21% | 43% | 6% |
| Plasmepsin II | 0.162 | [ 79]-[ 90], [ 93]-[103], [124]-[128] |
21% | 43% | 6% |
| HAP | 0.160 | [ 74]-[115], [163]-[190], [197]-[201], [287]-[295], [304]-[308] | 20% | 44% | 4% |
| Plasmepsin IV | 0.174 | [ 78]-[102], [390]-[395] |
20% | 40% | 4% |
| Plasmepsin V | 0.114 | [ 47]-[ 93], [269]-[336],[462]-[469], [472]-[486], [507]-[513] | 14% | 32% | 9% |
| Plasmepsin VI | 0.169 | [ 42]-[ 78], [129]-[170], [239]-[262], [353]-[362] |
18% | 40% | 4% |
| Plasmepsin VII | 0.098 | [ 38]-[ 73], [119]-[129], [234]-[261], [271]-[329], [385]-[418] | 13% | 40% | 4% |
| Plasmepsin VIII | 0.143 | [ 77]-[117], [119]-[126], [198]-[204] |
10% | 45% | 4% |
| Plasmepsin IX | -0.018 | [ 75]-[ 87], [ 89]-[225], [ 89]-[225], [427]-[508], [571]-[575], [595]-[627] | 10% | 38% | 3% |
| Plasmepsin X | 0.037 | [ 49]-[228], [278]-[283], [289]-[293], [303]-[312] |
17% | 35% | - |
Table 4: Identification of unfold ability, region of disorder segment in the sequence and the secondary structure predictions of each plasmepsin shown in table showing intrinsic protein disorder regions predicted by FoldIndex and GLOBPLOT 2 and unfold ability predicted by FoldIndex. In FoldIndex positive values represent proteins likely to be folded, and negative values represent those likely to be intrinsically unfolded. The percentage of alpha helix, beta sheet and Tm helix predicted using PSIPRED.
To the better understanding of the evolutionary relationships plasmepsins from P. falciparum, a phylogenetic analysis was performed by using MEGA 5.1 (Figure 3). The evolutionary history was evaluated based on the Neighbor-Joining method by bootstrap technique. This technique allows evaluating and judging the strength of branching pattern of tree. Also, the bootstrap value given at the node of each branch determines the probability of correct or incorrect relationship such that the value above 50% represents higher confidentiality of relationship and vice versa. The phylogenetic tree is separated into four distinct cluster groups with varied values where plasmepsin I, II, IV and HAP are present in single cluster. Cluster I contains plasmepsins, which are located in the acidic food vacuole and are active during the intra-erythrocytic phase of the life cycle. The sequence, structure and function of these proteins have more than 60% identity and fall together in a single cluster. The second cluster contains the plasmepsin IX and X, where these two proteins are known to express concurrently with plasmepsin I–IV, but their sequence identity is less than 30%. The third cluster contain plasmepsin VI, VII and VIII express within the sporogonic cycle in the mosquito and are functionally different from the other cluster of plasmepsins, Plasmepsin V forms a separate cluster indicating that this protein is functionally different from the other plasmepsins. It is an integral membrane protein present in the endoplasmic reticulum of the parasite and helps to export hundreds of proteins from the parasite to the host cell. These studies thus indicate that each cluster family is separated based upon its occurrence and localization.

Figure 3: The phylogenetic analysis of all the ten plasmepsin from P. falciparum which makes four different clusters.
For several years, the structure-based drug design of anti-malarial compounds targeting plasmepsin and its inhibitors has received much attention due to their potential biomedical use. The X-ray crystallographic structure of plasmepsin I, II, IV and HAP are well known but other plasmepsin structures are not known. The PDB structures of plasmepsin I-IV and HAP structure were downloaded from the PDB database. These four structures were superposed and highlights the structures having similar secondary structure fold when compared to that of eukaryotic aspartic proteases. The single aminoacid protein chain of these plasmepsins folded into a topologically similar way consisting of two major beta-hairpin in their domain region along with two catalytic aspartic acid residues present in the N- and C-terminal of the beta-hairpin structure. The PDB structure of plasmepsin II (1SME) with its disulfide linkage and active site residues is shown in Figure 4a.

Figure 4: a) The crystal structure of plasmepsin, where the blue color represents it’s active site region, active site residue ASP is marked and the magenta color represents disulfide [S] linkage region. b) The superposed structure of all the known plasmepsins where green, yellow, magenta and cyan represents plasmepsin I, II, IV and HAP respectively. The red color circle represents the cavity of the proteins. c) The superposed structure of HAP (green color) against the modeled structure were plasmepsin VI (cyan color), VII (yellow color), VIII (magenta color), IX (brown color), X (gray color). d) The superposed structure of plasmepsin V (green color) over the structure of Plasmodium vivax (red color) and the blue circle indicate the extra regions which not present in the structure of plasmepsin V from Plasmodium vivax.
The unknown structures of other plasmepsins (Plasmepsin V, VI, VII, VIII, IX and X) were predicted using SWISS-MODEL and PHYRE structure prediction server. Plasmepsin V was modeled based on the X-ray crystallographic structure of plasmepsin V from Plasmodium vivax that served as the template 4ZL4 [63]. The missing regions in the crystal structure of plasmepsin V of Plasmodium vivax were also modeled in the built structure of plasmepsin V from Plasmodium falciparum (Figure 4d). The other plasmepsins VI-X were modeled against the PDB structure of cathepsin D (4OBZ), the most closely related human aspartic protease. All the single chain modeled structures except plasmepsin V were further superimposed over the crystal structure of cathepsin D from Homo sapiens [64] (Figure 4c). Plasmepsin V was superposed against its homolog from Plasmodium vivax (Figure 4d) and we also superposed the structure of all known plasmepsins, plasmepsin I-IV, together (Figure 4b). The structure of all the superposed model showed a similar topological fold which represent that the ten plasmepsins comes into the same fold of pepsinlike superfamily.
The quality and reliability of modeled structure was checked by several structural assessment methods like Z-score, RMSD and Ramachandran plot. The Z-Score and the RMSD value of the modeled 3D structure were predicted using PROSA web server and Dali server and the results are tabulated in Table 6. The Dali server gives result as a pair of pre-computed structural neighbor, which has been sorted by Z-score and the Z-score lower than 2 are considered spurious. The RMSD has often been used to measure the quality of reproduction of a known (i.e., crystallographic) binding pose by a computational method and the RMSD value of each model were less than 1 indicate the best models. The Ramachandran plot provides an easy way to view the distribution of torsion angles of a protein structure [65]. It also provides an overview of allowed and disallowed regions of torsion angle values, serving as an important indicator of the quality of protein 3D structures. Based on the Ramachandran plot, almost all the plasmepsin models are having 90% residues in the allowed region, whereas plasmepsin V has 93% residues in their allowed region. This indicates the degree of correctness of modeling of the plasmepsin proteins. The models were further checked by VERIFY3D, the analysis revealed the number of amino acids have scored ≥ 0.2 in the 3D/1D profile and the result showed in Table 5. All the predicted structure validation depicted that plasmepsin V has a better modeled structure as compared to the other plasmepsin structures. This summary may be useful for biologists seeking a good crystallographic structure and aiming towards exploiting the pre modeled structures for docking analysis.
| Protein | Template | Sequence identity | RMSD Value | Z -score | Total number of buried outlier protein atoms | Overall quality factor | Averaged 3D-1D score ≥ 0.2 | Number of residues in favored region | Nunmberof residues in allowed region | Number of residues in outlier region |
|---|---|---|---|---|---|---|---|---|---|---|
| Plasmepsin V | 4ZL4 | 64% | 0.4 | 38.0 | 101 | 75.01 | 80.0% | 93.3% | 8.2% | 1.6% |
| Plasmepsin VI | 4obz | 39% | 0.5 | 39.6 | 46 | 61.483 | 79% | 91.94% | 3.4% | 3.4% |
| Plasmepsin VII | 4obz | 30% | 0.5 | 52.1 | 50 | 60 | 65% | 91.8% | 5.6% | 0.6% |
| Plasmepsin VIII | 4obz | 34% | 0.8 | 43.3 | 41 | 55.625 | 70% | 89.25% | 5.38% | 5.38% |
| Plasmepsin IX | 4obz | 35% | 0.8 | 43.5 | 50 | 61.12 | 75% | 91% | 6.25%) | 2.15%) |
| Plasmepsin X | 4obz | 35% | 0.8 | 35.0 | 58 | 58.075 | 74.54% | 90.1% | 7.7% | 3.3% |
Table 5: The checking and validation of protein structures during and after the model refinement of plasmepsin V- X using various software's available in SAVES. The RMSD value of each model is predicted from DALI server, Z-score predicted using PROSA web server. The total number of outlier protein atoms are predicted from PROVE and averaged 3D-ID score calculated from VERIFY 3D using the modeled structures. The number of residues in the favored region, allowed region and outlier region is checked using Ramachandran plot.
All the plasmepsins of P. falciparum are aspartic proteases, which constitute one of the major protease subclasses. They further distinguish each subclass based on its structural homology. Most of the aspartic proteases, including the plasmepsins and cathepsin D, are members of the pepsin family. Most of the known aspartic proteases have a well- defined subsite pocket to inhibit its substrate pepstatin. These subsites are located on both side of the catalytic site, found only in eukaryotes. On the basis of X-ray crystallography, it is well known that pepstatin binds to plasmepsin I-IV in its enzyme binding subsites, which is localized in the extended beta-strand conformation. The structures also contain a beta-hairpin turn, which helps to interact with substrate and inhibitors by covering the binding cavity. The substrate binding cavity and its interacting residues are similar in plasmepsin I- IV and HAP protein. The co-crystal structure of plasmepsin II (PDB ID: 1W6I) is of 329 amino acid residue and its catalytic binding sites contain 1 non-polar residue (Val 78), 5 polar residues (Gly 36, Asn 76, Ser 79, Tyr 192 and Ser 218) and 2 negatively charged residues (Asp 34 and Asp 214) (Figure 5a). The ligand pepstatin formed five H-bond with the catalytic residues (Figure 5b). The other amino acid residues Ser 37, Tyr77, Gly 216, Thr 217, Phe 241, Leu 242 and Ile 290 were also involved in the hydrophobic interactions with pepstatin.

Figure 5: a) The crystallographic structure of plasmepsin II in complex with pepstatin where the blue dot represents the hydrogen bonding and the green ball and stick molecules indicate pepstatin. b) Intermolecular interactions in docked complex of plasmepsin 2 and pepstatin where green colored dotted line denotes hydrogen bond formation between the ligand and protein. (PDB ID: 1W6I).
The modeled structures of all the plasmepsins were superposed over plasmepsin II, which was further, used for docking studies. The binding sites of each model were predicted based on the alignment of the binding residues of plasmepsin II. An automated docking tool, Autodock Vina that works by Lamarckian Genetic Algorithm, performed molecular docking studies [66]. Their docking score between ligand and model predicts the strength and the binding activity of a binding complex. The docking score of pepstatin with each plasmepsin model were calculated (Table 6). The docking score of pepstatin with plasmepsin I-VI and plasmepsin VII-X were below a score of -7 affinity, whereas HAP and plasmepsin VI have a binding score of -6.5 and -6.6 respectively. The least binding score was observed -5.1 with plasmepsin V indicating that plasmepsin V have a low binding affinity towards pepstatin. The least binding of plasmepsin V with pepstatin as compared to other plasmepsins might be because of low volume of the cavity due to the loop located at the cleft of the binding pocket. We further designed some small molecules similar to the pepstatin and used them as a ligand for docking studies. The shape of the binding pocket of plasmepsin V (Figure 6a) was observed to complement the shape or pose of the ligand and the grid was made accordingly for the docking studies (Figure 6b). Compared to the peptide molecules the designed small molecules showed more binding affinity towards plasmepsin V (Table 7). Almost all the molecules except M2 produced a docking score less than -8 kcal/ mol and M2 showed an affinity of -7.7 kcal/mol. The molecule M9 showed lowest binding score with two hydrogen bonding between GLY 367 and ASP 11. The other molecules M5 and M7 with model have hydrogen bond between GLY 367. The possible interacting residue of plasmepsin V model with each ligands were shown in Figure 7 and Table 7. These results might be helpful for structure based inhibitor designing for plasmepsin V. This study opens the wide area of research focusing on synthesis of these molecules for in vivo studies which may also lead to design new inhibitors for plasmepsin V.
| Sl. No | Protein Name | Binding energy | Residues in the hydrophobic cavity |
|---|---|---|---|
| 1 | Plasmepsin I | -7.3 | Phe101, Gly115, Ile120, Ile30,Met13, Thr222, Val289, Ile287 |
| 2 | Plasmepsin II | -7.1 | Ile123, Tyr77, Ser79, Val78, Phe244, Phe294, Ile290, Ile212, Gly213, Tyr192, Ile123 |
| 3 | HAP | -6.8 | Leu73, Ile80, Met140, Trp34, Leu20, Phe15, Ala10, Trp94, Ser115, Tyr112 |
| 4 | Plasmepsin IV | -7.2 | Gly78, Typ77, Ser76, Ile294, Val292 |
| 5 | Plasmepsin V | -5.6 | Tyr341, Leu363, Asp365, Val486, Asn485 |
| 6 | Plasmepsin VI | -6.9 | Gly120, Tyr167, Thr169, Ala318, Arg396, Val334, Leu392 |
| 7 | Plasmepsin VII | -7.8 | Ile205, Trp202, Ile194, Ser181, Phe85, Thr327, Thr418, Ile322 |
| 8 | Plasmepsin VIII | -7.0 | Ile123, Ile359, Trp67, Leu170, Tyr239, Gly272, Val258, Tyr57, Lys53, Phe125 |
| 9 | Plasmepsin IX | -7.1 | Ile244, Asp246, Tyr409, Asp496, Ile577, Leu220, Tyr293, Phe333, Phe291 |
| 10 | Plasmepsin X | -7.1 | Ile264, Phe311, Arg244, Ile354 |
Table 6: The ligand binding energy and its interacting residues against the structure of plasmepsins after docking studies.

Figure 6: a) The modeled structure of plasmepsin V with its ligand M9 docked in its binding pocket. b) The grid defined for binding pocket of modeled plasmepsin V.
| Number | Molecule | Binding affinity (skcal/mol) | Dist from best mode rmsdl.b. | Dist from best mode rmsdu.b. | Interacting residues and nearby residues |
|---|---|---|---|---|---|
| M1 |  |
-8.1 | 0.938 | 5.987 | ASP 365, GLY 367, CYS178, GLU 179, TYR177, LEU 218 |
| M2 |  |
-7.7 | 4.057 | 7.547 | THR369, SER 368, ALA98, TYR99, ILE 116, PHE 219, TYR177, GLU 179, CYS 178 |
| M3 |  |
-9.1 | 3.713 | 6.837 | ILE 491, GLY 397, TYR99, ILE 116, LEU218, CYS178, TYR 177 |
| M4 |  |
-8.1 | 0.000 | 0.000 | GLY367, ILE 116, GLU 179, TYR 177, LEU 218, CYS 178, TYR 177 |
| M5 |  |
-8.6 | 3.402 | 5.198 | GLY 367, HIS372, CYS 178, GLU 179, TYR 177, GLY 367, VAL 227 |
| M6 |  |
-8.3 | 4.057 | 7.547 | TYR 99, ASP118, TYR 177, LEU 218, HIS 221 |
| M7 |  |
-8.2 | 2.335 | 5.571 | TYR 99, GLY 367, CYS 178, GLU 179, TYR 177, LEU 218, ILE 116 |
| M8 | 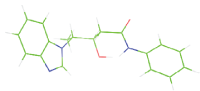 |
-8.2 | 0.000 | 0.000 | GLY367, CYS 178, GLU 179, TYR 177, ILE 116, TYR 99 |
| M9 | 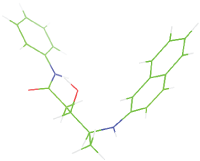 |
-9.2 | 0.880 | 4.894 | TYR 99, GLY 367, GLU 179, LEU 218, ASP 11, VAL 227 |
Table 7: The designed small molecule along with its binding energy towards plasmepsin V shown in table. The color code is based on the atoms presents in the molecule where green color represents carbon atom, gray color represents hydrogen atom, blue represents nitrogen and red color represents oxygen atoms.

Figure 7: The best docking conformation of molecules (M1-M9) and the nearby residues of modeled plasmepsin V. The ball and stick structure represents ligand, stick represents the residues interacting with ligands and green dots represents hydrogen bonding.
The overall study of plasmepsins and the experimentally known results indicate that these proteins play an extensive role in the survival of parasite inside the host cell. Plasmepsin I–IV are situated in the acidic food vacuole and are active only during the intra-erythrocytic phase of the life cycle. Since, they are involved in the degradation of host hemoglobin; their inhibition is considered to be a good anti-malarial strategy. Plasmepsin VI-VIII is expressed within the sporogonic cycle in the mosquito, but not much research has been done on their functional part. Hence, these plasmepsins expressed in the intra-erythrocytic stage may not be suitable for designing drugs against malaria at this point of time. Similarly, plasmepsin IX and X are expressed concurrently with plasmepsin I–IV, but are not transported to the food vacuole. Their large molecular weight, disordered regions and their inefficiency to build a properly folded structure are major obstructions for designing good inhibitors against them. Plasmepsin V present in the endoplasmic reticulum of the parasite helps to export hundreds of proteins into the host cell to remodel its erythrocyte surface. The recent preliminary docking result in this article helps in better understanding of plasmepsins and also unfastens wide area for research focusing on structural characterization and design & synthesis of small molecule that are potent candidate for anti-malarial therapy with respect to Plasmepsin inhibition.
The present study is focused on the In-silico characterization and structural investigation of plasmepsins from Plasmodium falciparum. The domain prediction revealed that all the plasmepsins contain a eukaryotic aspartic protease domain and have conserved active site pattern in their N- and C-terminal. A physicochemical characterization was performed by computing theoretical isoelectric point (pI), molecular weight, extinction coefficient and instability index together with the prediction of disulfide linkages, motif profiles, sub-cellular localization and disordered regions using various servers. The fold of modeled structures was compared with the known three dimensional structure plasmepsins and the models were validated using Ramachandran’s map, PROCHECK and WHAT IF. The docking studies of plasmepsin were done with its known inhibitor pepstatin. We also designed some small molecule inhibitors for plasmepsin V which may serve as a good foundation for designing new inhibitors for plasmepsins Table 8.
| Tools used | Website | Use |
|---|---|---|
| NCBI | http://www.ncbi.nlm.nih.gov/ | Sequence retrieval. |
| PlasmoDB | http://plasmodb.org/plasmo/ | Database of the Plasmodium falciparum genome sequencing consortium. |
| CLUSTAL X | Stand-alone software | It provides an integrated environment for performing multiple sequence and profile alignments and analysing the results, |
| SMART | http://smart.embl-heidelberg.de/ | SMART used to explore domain architectures. |
| Pfam database | http://pfam.xfam.org/ | Look at the domain organisation of a protein sequence. |
| PRED-CLASS server | http://athina.biol.uoa.gr/PRED-CLASS/ | Classification of proteins into one of four possible classes like the membrane protein, globular protein, fibrous protein and the mixed (fibrous and globular) protein. |
| ProtParam | http://web.expasy.org/protparam/ | It compute the various physical and chemical parameters of protein |
| PROSITE | http://prosite.expasy.org/ | It describing protein domains, families and functional sites |
| HMM-TM: | http://aias.biol.uoa.gr/HMM-TM/ | It predicts the transmembrane regions of alpha-helical membrane proteins, trained on crystallographically solved data. |
| NetNGlyc 1.0 Server | http://www.cbs.dtu.dk/services/NetNGlyc/ | The NetOglyc server produces neural network predictions of mucin type GalNAc O-glycosylation sites in mammalian proteins. |
| FoldIndex | http://bioportal.weizmann.ac.il/fldbin/findex | Tool to predict whether a given protein sequence is intrinsically unfolded, which is based on the average residue hydrophobicity and net charge of the sequence. |
| PHYRE2 Protein Fold Recognition Server | http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index | Used for protein modeling. |
| MEGA software | http://www.megasoftware.net/ (Stand-alone software) |
Used for conducting statistical analysis of molecular evolution and for constructing phylogenetic trees. |
| DISULFIND | http://disulfind.dsi.unifi.it/ | Predicting the disulfide bonding state of cysteines and their disulfide connectivity starting from sequence alone |
| DiANNA web server | http://clavius.bc.edu/~clotelab/DiANNA/ | Software for Cysteine state and Disulfide Bond partner prediction |
| PSIPRED | http://bioinf.cs.ucl.ac.uk/psipred/ | Used for protein secondary structural prediction |
| Phobius prediction | http://phobius.sbc.su.se/ | A combined transmembrane topology and signal peptide predictor |
| GLOBPLOT 2 | http://globplot.embl.de/ | Exploring protein sequences for globularity and disorder |
| PyMOL | https://www.pymol.org/ | Molecular visualization |
| Swiss PDB viewer | http://spdbv.vital-it.ch/ | Molecular visualization |
| DALI server | ekhidna.biocenter.helsinki.fi/dali_server/start | Comparing protein structures in 3D |
| RAMPAGE server | RAMPAGE server | Used for Ramachandran Plot Analysis |
| AutoDock Vina | Stand-alone software | Automated docking software designed to predict how small molecules, such as substrates or drug candidates, bind to a receptor of known 3D structure. |
| PROCHECK | http://www.ebi.ac.uk/thornton-srv/software/PROCHECK/ | It checks the stereo chemical quality of a protein structure |
| PROVE software’s | http://services.mbi.ucla.edu/ | The volume-based structure validation procedures are implemented in the program PROVE |
| SuperPose | http://wishart.biology.ualberta.ca/SuperPose/ | Used for the superposition of two or more structures using a modified quaternion approach |
| ERRAT | http://services.mbi.ucla.edu/ERRAT/ | This method is for differentiating between correctly and incorrectly determined regions of protein structures based on characteristic atomic interaction is described. |
Table 8: Lists out the different software’s used for the protein modelling and bioinformatics web servers used for in-silico analysis.
The authors thank Department of Biotechnology (DBT) for RA fellowship to Divya Nair and IIT Gandhinagar for research facilities and infrastructure. We also thank Dr. Kirubakaran, IIT Gandhinagar for her inputs in writing this manuscript.