Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2016) Volume 9, Issue 11
This paper discusses the sequence/structure relation. The core question concerns the degree to which similar sequences produce similar structures and vice versa. A mechanism by which similar sequences may result in dissimilar structures is proposed, based on the Fuzzy Oil Drop (FOD) model in which structural similarity is estimated by analyzing the protein’s hydrophobic core. We show that local changes in amino acid sequences, in addition to producing local structural alterations at the substitution site, may also change the shape of the hydrophobic core, significantly affecting the overall tertiary conformation of the protein. Our analysis focuses on four sets of proteins: 1) Pair of designer proteins with specially prepared sequences; 2) Pair of natural proteins modified (mutated) to converge to a point of high-level sequence identity while retaining their respective wild-type tertiary folds; 3) Pair of natural proteins with common ancestry but with differing structures and biological profiles shaped by divergent evolution; and 4) Pair of natural proteins of high structural similarity with no sequence similarity and different biological function.
Keywords: Structure comparison; Structure alignment; Structural differences; Structure prediction; Evolution; Protein folding; Hydrophobicity
The presented analysis concerns the well-known problem of correlating the protein’s amino acid sequence with its 3D structure [1- 4]. The search for algorithms which can be used to translate the former into the latter is a fundamental problem in proteomics [5,6] and often yields useful insight into the specific properties of individual proteins [7].
A classic example of this phenomenon is the group of structures referred to as immunoglobulin-like domains. Such domains are present in all immunoglobulins (where they determine their function) but are also encountered in enzymes and transport proteins [8- 10]. Immunoglobulins exhibit high structural similarity, adopting characteristic “sandwich” conformations with rather low sequence similarity. Even among immunoglobulins domains the λ and κ sequences are identified. Of course, the diversity of proteins which are not immunoglobulins but which do contain immunoglobulin-like domains is even greater [10].
In addition to the above, studies have revealed cases where very similar sequences produce significantly different structural forms [11]. For example, the KGVVPQLVK sequence generates a classic β-twist in 1PKY but adopts a helical conformation in 1IAL [12-14]. The three 7-residue sequences which also share this property of different secondary structure for identical sequences are given in Jacoboni et al. [15].
Conservative hydropathic identity at geometrically equivalent positions is the object of analysis in Krissinel [16]. Our work focuses on structural differences in four pairs of proteins: 1) Pair of designer proteins with specially prepared sequences; 2) Pair of natural proteins modified (mutated) to converge to a point of high-level sequence identity while retaining their respective wild-type tertiary folds; 3) Pair of natural proteins with common ancestry but with differing structures and biological profiles shaped by divergent evolution; and 4) Pair of natural proteins of high structural similarity with no sequence similarity and different biological function.
In attempting to show the role of hydrophobic core structure in structural stabilization we refer to the Fuzzy Oil Drop (FOD) model, which predicts the 3D conformation of the target protein by simulating the emergence of a hydrophobic core. While our research has identified some interesting correlations, generalizing them remains an open issue: in order to determine whether the presented results may, in fact, be generalized we need to process a much larger database of proteins.
Data
The presented analysis concerns four sets of two proteins each. The first set comprises two de novo designed proteins with a sequential similarity of 88% but with differing 3D structures. The second describes two natural proteins which are modified (mutated) in a stepwise fashion in order to align their sequences while preserving structural differentiation (helix-to-Beta). The third set includes two natural homologues with common ancestry. The fourth one discusses pair of natural proteins of high structural similarity with no sequence similarity and different biological function.
de novo design
Two de novo designed proteins, 2JWU and 2JWS, also referred to as GA88 and GB88 share 88% sequence identity but different folds [17]. The geometries of the seven nonidentical residues (of 56 total) provide insight into the structural basis for switching between 3-α and α/β conformations. Further mutation of a subset of these no identities, guided by the GA88 and GB88 structures, leads to proteins with even higher levels of sequence identity (95%) and differing folds. Thus, conformational switching to an alternative monomeric fold of comparable stability can be effected with just a handful of mutations in a small protein. This result has implications for understanding not only the folding code but also the evolution of new folds. The CATH [18] classification for these two proteins is as follows: 1.10.8.40- Mainly Alpha Orthogonal Bundle for 2JWS and 3.10.20.10.-R-Alpha Beta Roll for 2JWU.
Wild type proteins with aim-oriented mutations
Two proteins: G311 (1ZXG) and A219 (1ZXH) are modified versions of wild-type proteins designated G and A (IgG binding domains, source: Staphylococcus aureus for 1ZXG and Streptococcus sp. for 1ZXH) respectively [19]. The series of mutations aimed at achieving a high level of sequence identity while preserving wild-type 3D structures. Both proteins (G311 vs. G and A219 vs. A) represent backbone RMS-D 1.4 Å, maintaining wild-type secondary structures: α/β for G311 and helical for A219. The final sequence identity of both modified proteins is on the level of 59%. All relevant data was taken from a paper describing experimental results of protein modifications [19].
Homologous proteins
The next pair comprises two proteins with common ancestry but different folds and functions: 2PIJ (Pfl 6 BETA) (source: Pseudomonas fluorescens) Cro protein from prophage pfl 6 in P. fluorescens pf-5 [20] and 3BD1 (Xfaso 1) Cro protein from putative prophage element X. fastidiosa strain ann-1 [20] (source: Xylella fastidiosa). Both proteins share an identical CATH classification: 1.10.260.40-Mainly Alpha Orthogonal Bundle.
The differences between homologous proteins are due to evolutionary pressure. In the presented case the sequence identity is 40%, yet the α-helix is replaced by a β-sheet in the C-terminal region spanning approximately 25 residues. According to Roessler et al. [20], sedimentation analysis suggests a correlation between helix-to-sheet conversions, along with strengthened dimerization.
Unrelated proteins of similar fold: Two proteins not related in respect neither to evolution nor to function representing however the common fold classified by CATH as 3.40.50.360–3CHY (signal transduction protein) [21] and 3.40.50.2300–1RCF (flavodoxin) [22] both of 3-layer (aba) sandwich form with no sequence similarity (about 19%).
Introduction to the Fuzzy Oil Drop (FOD) model
The Fuzzy Oil Drop (FOD) model is a modification of the previously described oil drop model which asserts that hydrophobic residues tend to migrate to the center of the protein body while hydrophilic residues are exposed on its surface [23,24]. The FOD replaces the binary discrete model [25] with a continuous function peaking at the center of the molecule [23], which causes hydrophobicity density values to decrease along with distance from the center, reaching zero on the molecular surface. The idealized, theoretical hydrophobicity distribution is expressed by 3D Gauss function. The size of molecule shall be expressed by sigma parameters for Gauss function. The characteristics of this function allows represent the hydrophobicity distribution with maximum in the center of ellipsoid with decrease together with the increase of distance versus the center reaching zero level in the distance equal to 3sigma in any direction. This idealized distribution ensures high solubility since the entire ellipsoid is covered by the hydrophilic shell.
On the other hand the actual distribution of hydrophobicity density observed in a protein molecule depends on inter-chain interactions, which, in turn, depend on the intrinsic hydrophobicity of each amino acid. Intrinsic hydrophobicity can be determined by experimental studies or theoretical reasoning– our work bases on the scale published in Kalinowska et al. [24] while the force of hydrophobic interactions has been calculated using other scales as it was shown in Kalinowska et al. [24]. For each amino acid j (or, more accurately, for each effective atom) the sum of interactions with its neighbors is computed and subsequently normalized by dividing it by the number of elementary interactions (following the function proposed in Levitt [26].
The two hydrophobicity density distribution profiles: the expected (T) and observed (O) distribution an be compared quantitatively. Quantitative expressing of the differences between the expected (T) and observed (O) distribution is possible using the Kullback-Leibler divergence entropy formula [27]:
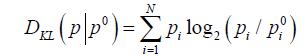
The value of DKL expresses the distance between the observed (p) and target (p0) distributions, the latter of which is given by the 3D Gaussian (T). The observed distribution (p) is referred to as O.
For the sake of simplicity, we introduce the following notation:
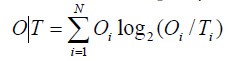
Since DKL is a measure of entropy it must be compared to a reference value. In order to facilitate meaningful comparisons, we have introduced another opposite boundary distribution (referred to as “uniform” or R) which corresponds to a situation where each effective atom possesses the same hydrophobicity density (1/N, where N is the number of residues in the chain). This distribution is deprived of any form of hydrophobicity concentration at any point in the protein body:
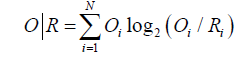
Comparing O|T and O|R tells us whether the given protein (O) more closely approximates the theoretical (T) or uniform (R) distribution. Proteins for which O|T>O|R are regarded as lacking a prominent hydrophobic core. To further simplify matters we introduced the following Relative Distance (RD) criterion:
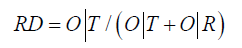
RD<0.5 is understood to indicate the presence of a hydrophobic core. Figure 1 presents a graphical representation of RD values, restricted (for simplicity) to a single dimension.

Figure 1: Graphical representation of fuzzy oil drop model hydrophobicity distributions obtained for a hypothetical protein reduced to a single dimension for simplicity. A) Theorized Gaussian distribution (blue) while the chart C corresponds to the uniform distribution (green). Actually observed (red) hydrophobicity density distribution in the target protein B, while its corresponding value of RD (relative distance), and in D is marked on the horizontal axis with a red diamond. According to the fuzzy oil drop model this protein does not contain a well-defined hydrophobic core, because its RD value, equal to 0.619, is above the 0.5 threshold (or-generally-closer to R than T).
DKL (as well as O|T, O|R and RD) may be calculated for specific structural units (protein complex, single molecule, single chain, selected domain etc.) In such cases the bounding ellipsoid is restricted to the selected fragment of the protein. It is also possible to determine the status of polypeptide chain fragments within the context of a given ellipsoid. This procedure requires prior normalization of O|T and O|R values describing the analyzed fragment.
RD can be calculated for entire units (protein, chain, domain) and for selected fragment (following normalization of Ti and Oi values of the fragment under consideration).
The above procedure will be applied in the analysis of proteins described in this paper. By restricting our analysis to individual fragments, we can determine whether a given fragment participates in the formation of a hydrophobic core. In particular, fragments of chain representing well defined secondary folds which satisfy RD<0.5 are thought to contribute to structural stabilization, while fragments for which RD≥ 0.5 are less stable. Such fragments, if present on the surface of the protein, may potentially form complexation sites. The fragments of chains are defined by their secondary structure. Identification of secondary structural folds and the composition of protein domains follow the CATH [18] and PDBsum [28] classifications. Likewise, interdomain/ inter-chain contacts have been identified on the basis of the PDBsum distance criteria [28].
The graphic presentation of RD interpretation is shown in Figure 1.
The OORF system of RD calculation uses the method from ORF calculation in DNA analysis. OORF stands from Overlapped Open Reading Frame. The window of declared size (10 aa in our analysis) is taken as the fragment, the RD value is calculated. For example fragment 1-10 gets described by its RD value. Then the next window (2-11 aa) is taken for RD calculation. The RD value for each window requires prior normalization (the sum of Ti and Oi belonging to the window shall be equal to 1.0). This form of calculation makes possible characteristics of entire chain regardless the secondary structure.
The detailed description of the FOD model is available in the paper recently published [29].
de novo designed proteins
According to results given in Table 1 the structure of 2JWS (GA88) is consistent with the model both as a whole and in its packed section (without the N-terminal fragment 1-7 which was eliminated from calculation since the FOD model works well with globular proteins). This operation does not affect the status of helical folds.
| SECONDARY FRAGMENT | O|T | O|R | RD | |
|---|---|---|---|---|
| 2JWU (GB88) | COMPLETE MOLECULE | 0.126 | 0.249 | 0.335 |
| B 1-9 | 0.052 | 0.157 | 0.249 | |
| B 12-20 | 0.196 | 0.248 | 0.441 | |
| Mut–4 | H 22-37 | 0.146 | 0.203 | 0.419 |
| L 38-41 | 0.078 | 0.069 | 0.533 | |
| Mut–1 | B 42-46 | 0.203 | 0.160 | 0.558 |
| Mut–1 | B 50-56 | 0.066 | 0.180 | 0.268 |
| 2JWS (GA88) | COMPLETE MOLECULE | 0.182 | 0.308 | 0.371 |
| Unstructure 1-7 | 0.235 | 0.241 | 0.493 | |
| Mut–1 | H 8-24 | 0.260 | 0.322 | 0.447 |
| Mut–2 | H 26-35 | 0.055 | 0.251 | 0.181 |
| Mut–3 | H 38-52 | 0.129 | 0.179 | 0.418 |
| 2JWS (GA88) 8-56 | COMPLETE DOMAIN | 0.170 | 0.277 | 0.380 |
| Mut–1 | H 8-24 | 0.213 | 0.328 | 0.394 |
| Mut–2 | H 26-35 | 0.078 | 0.250 | 0.238 |
| Mut–3 | H 38-52 | 0.161 | 0.175 | 0.479 |
Table 1: Properties of selected proteins from the point of view of the fuzzy oil drop model. O|T values indicate the distance between the observed and theoretical distribution while O|R values show the corresponding distance between the observed and uniform distribution. RD values are in relation to the idealized (theoretical) distribution. Each parameter is listed for complete molecules and for their individual folds. The leftmost column also includes information on sequential discrepancies between pairs of molecules. Mut-N indicates an exchange and the number of affected residues (2JWS vs. 2JWU) in each fragment. The bottom part of the table shows results for the packed domain of 2JWS (without the unstructured N-terminal fragment). B stands for β-type structures while H indicates a right-handed α-helix.
In 2JWU (GB88) four β-folds can be distinguished, in addition to a single helix. This molecule also contains a loop (38-41). The β-fragment at 42-46 and the loop both diverge from the model even though in GA88 the same residues form parts of an accordant helix. The fragment at 38-46 is characterized by higher-than-expected hydrophobicity density (Figure 2A). Since this fragment is exposed on the protein surface (expected hydrophobicity is low), it may be responsible for possible forming complexes with other proteins which also expose hydrophobic areas on their surface.

Figure 2: Hydrophobicity density distribution profiles for two molecules (de novo design) differing with respect to their secondary structure: A, B-2JWS (GA88), C, D-2JWU (GB88). Theoretical hydrophobicity profile is plotted in blue while that of observed hydrophobicity is red. Vertical green lines localize positions of mutations. Orange shades under the profiles indicate positions of hydrophobic core, understood as groups of residues with peak values of both hydrophobicity distributions. Figures 2B and 2C present RD values of both proteins calculated in the OORF (Overlapped Open Reading Frame) system with 10 aa window size. Red and yellow fragments visible on those figures denote locations of secondary structure fragments: α-helices and β-sheets, respectively.
The consistently high accordance of 2JWS–both as a whole and when subdivided into folds–suggest a relative lack of local deformations. It may be speculated that as predicted by the FOD model, this molecule is highly water-soluble with low tendency to interact with any ligand molecule.
Figures 2A and 2D present the hydrophobicity density profiles for both proteins, showing the values ascribed to each residue in the polypeptide chain. The distinguished fragments satisfy the condition of high expected and high observed hydrophobicity in both molecules. From the point of view of the model both molecules contain well-defined hydrophobic cores. One shall notice that the FOD model identifies the central part of molecule as the hydrophobic core together with the shell of intermediate coat including the exposed surface of expected hydrophobicity close to zero (hydrophilic surface). The co-existence of these two parts makes the hydrophobic core complete as protected and isolated against immediate contact with water environment. The identification of residues recognized as hydrophobic core members is based on the high expected and high observed hydrophobicity. Residues following this condition are recognized as responsible for hydrophobic core construction.
The profiles shown in Figures 2B and 2C (OORF distributions) reveal significant differences pointing different fragments of low RD values suggesting high accordance between expected and observed distributions in both proteins.
The observations listed above seem to support the conclusion that both proteins are structurally different in terms of their hydrophobic cores. The construction of hydrophobic core in 2JWS is generated by central part of polypeptide chain, comprising two fragments (Figure 2A) while in 2JWU, it requires three separate fragments to participate in core generation (Figure 2D).
At this point it might be interesting to speculate about the progress of the folding process in each of these two cases. In 2JWS the hydrophobic core nucleates near the center of the chain, with the remaining sections aligning themselves to the emerging core. While in 2JWU the nucleation is mainly constructed by N- and C-terminal fragments with the participation also of central fragment of the chain.
Figure 3 presents a comparison of the status of each fold in both proteins, indicating mutations and the correspondence between the observed and theoretical hydrophobicity density in each fold. The makeup of the hydrophobic core is also depicted.

Figure 3: 3D protein structure model. A) 2JWU, B) 2JWS.
In summary, the introduction of seven mutations (G24A, I25T, I30F, I33Y, L45Y, I49T, L50K–with 2JWS serving as the reference strain) results in a far higher concentration of hydrophobic residues in 2JWS. This enlarges the hydrophobic core which is formed by the central fragment of the polypeptide chain. Unlike 2JWS, in 2JWU the core is made up of three separate fragments, including one that forms part of the shell (with lower hydrophobicity density). Substitutions at G42A, I25T, I30F, I33Y, L45Y result in the appearance of a long fragment which forms part of the hydrophobic core, while the presence of Y, T and K in the C-terminal fragment of 2JWU causes a hydrophobicity density gradient to emerge in the surface zone where hydrophilic residues appear, in accordance with the theoretical model. Figure 3 shows clearly the influence of mutations since the residues changed concern the positions of the central part in 2JWU. The location of these residues in 2JWU is rather distributed. In consequence different fragments of the chain participate in hydrophobic core formation and one fragment (the β-structural fragment) appears to represent the hydrophobicity density distribution discordant versus the idealized one.
Wild type proteins with aim-oriented mutations
The results listed in Table 2 indicate very high agreement with the FOD model in two compared proteins: 1ZXH and 1ZXG–two IgG binding domains. Additionally, each secondary structure (including loops) remains consistent with structural predictions provided by the model.
| SECONDARY FRAGMENT | O|T | O|R | RD | |
|---|---|---|---|---|
| A219 (1ZXH) | COMPLETE MOLECULE | 0.085 | 0.189 | 0.310 |
| Mut 1 | B 1-9 | 0.111 | 0.124 | 0.471 |
| Mut 4 | B 12-20 | 0.059 | 0.134 | 0.305 |
| Mut 6 | H 22-39 | 0.079 | 0.173 | 0.313 |
| Mut 3 | B 42-44 | 0.101 | 0.279 | 0.266 |
| Mut 4 | L 45-49 | 0.067 | 0.073 | 0.478 |
| Mut 3 | B 50-53 | 0.255 | 0.180 | 0.056 |
| B 54-55 | 0.039 | 0.160 | 0.197 | |
| G311 (1ZXG) | COMPLETE MOLECULE | 0.095 | 0.288 | 0.248 |
| L 1-7 | 0.087 | 0.196 | 0.308 | |
| Mut 2 | H 8-18 | 0.079 | 0.308 | 0.205 |
| Mut 5 | L 19-24 | 0.039 | 0.177 | 0.183 |
| Mut 2 | H 25-37 | 0.070 | 0.262 | 0.211 |
| Mut 11 | H 40-55 | 0.069 | 0.218 | 0.242 |
| L 56-59 | 0.100 | 0.493 | 0.168 |
Table 2: Properties of selected proteins from the point of view of the fuzzy oil drop model. O|T values indicate the distance between the observed and theoretical distribution while O|R values show the corresponding distance between the observed and uniform distribution. RD values are in relation to the idealized (theoretical) distribution. Each parameter is listed for complete molecules and for their individual folds. The leftmost column also includes information on sequential discrepancies between pairs of molecules. Mut-N indicates an exchange and the number of affected residues in each fragment.
Considering the large set of molecules analyzed using the FOD model we can conclude that the presented proteins are among the most accordant in the entire set, as indicated by their RD values (so far RD=0.38 for the immunoglobulin-like domain in titin (1TIT) was found to be the lowest). The structure of the hydrophobic core, which is understood as the entire tertiary conformation of the protein (including the core itself and its hydrophilic sheath) remains highly consistent with theoretical predictions, as shown in Figures 4A and 4B. Figures 4C and 4D illustrate the agreement between theoretical and observed hydrophobicity density distributions, with correlation coefficients of 0.847 and 0.784 for G311 and A219 respectively. The figures also reveal highly hydrophobic (core) and hydrophilic (surface) residues, whose placement can be seen in Figure 5A. Finally, Figure 5B marks the loci of point mutations–though the affected residues do not clearly belong either to the core or to the hydrophilic sheath. Figure 5 visualizes the positions of mutations and their influence on the hydrophobic core rearrangement.

Figure 4: Hydrophobicity density distributions and relations between them: (A and B) G311, (C and D) A219. Red, pointing upwards triangles mark residues accordant with the model and exhibiting high values of both kinds of hydrophobicity. Blue, pointing downwards triangles denote residues also accordant with the models, however presenting low hydrophobicity qualities. Cutoffs of 0.03 and 0.01 were chosen arbitrarily. Both proteins are highly accordant with the model, which also causes high correlation between their hydrophobicity distributions: 0.85 and 0.78 respectively.

Figure 5: 3D presentation of A219 and G311 molecules with highlighted residues.
Analysis of results for G311 and A219 indicates that tertiary structural stabilization (by hydrophobic core) appears to be dependent on a proper distribution of hydrophobicity density, ensuring the presence of a highly hydrophobic core as well as the encapsulating hydrophilic sheath, with near-zero hydrophobicity density values on its surface. Unfortunately the authors of the cited experimental work [17] do not report on the relation between the introduced mutations and the proteins’ capability to bind immunoglobulins. From the point of view of the FOD model, however, the mutated molecules should be less prone to complexation than their wild-type counterparts. This supposition follows from the observed good agreement between the theoretical and observed hydrophobicity density distributions–note that complexation sites are typically characterized by marked differences between both profiles (theoretical and observed). According to the model, a protein which only exposes hydrophilic residues on its surface should be highly soluble and incapable of interacting with any ligands other than dissolved ions. This phenomenon is evidenced by antifreeze and down-hill proteins, which exhibit near-perfect accordance with the theoretical hydrophobicity density distribution [30,31]. The role of these proteins is to be well soluble without any specific interaction with any molecules from environment except water to not allow the ice-structuralization of water.
Homologous proteins
Two proteins of common ancestry: 2PIJ and 3BD1 are characterized in Table 3. Both of them represent well defined hydrophobic core (distribution of observed hydrophobicity density is similar to expected one–RD below 0.5). Two secondary structural fragments were recognized as locally discordant in 2PIJ and one in 3BD1.
| SECONDARY FRAGMENT | O|T | O|R | RD | |
|---|---|---|---|---|
| 2PIJ | COMPLETE MOLECULE | 0.118 | 0.123 | 0.489 |
| B 2-5 | 0.088 | 0.161 | 0.356 | |
| H-6-13 | 0.023 | 0.194 | 0.105 | |
| H 15-24 | 0.077 | 0.034 | 0.689 | |
| H 26-36 | 0.036 | 0.044 | 0.453 | |
| B 39-45 | 0.105 | 0.178 | 0.370 | |
| B 49-55 | 0.077 | 0.046 | 0.625 | |
| 3BD1 | COMPLETE MOLECULE | 0.065 | 0.085 | 0.433 |
| A H 2-12 | 0.042 | 0.057 | 0.420 | |
| H 13-22 | 0.025 | 0.014 | 0.646 | |
| H 24-35 | 0.030 | 0.098 | 0.236 | |
| L, P | H 38-41 | 0.035 | 0.058 | 0.378 |
| P | H 42-49 | 0.030 | 0.067 | 0.309 |
| L | H 54-59 | 0.047 | 0.070 | 0.403 |
| L 50-53 | 0.014 | 0.091 | 0.135 |
Table 3: Comparison of fuzzy oil drop parameters for homologous proteins. O|T, O|R and RD values indicate the status of each molecule and each of its secondary structural fragments. “L” and “P” codes appearing in the leftmost column indicate involvement in ligand binding and protein complexation respectively. H and B denote helical and β-structural secondary fragments respectively.
Analysis of RD values calculated using the OORF system for two homologous proteins reveals differences in the structure of their hydrophobic cores. In 3BD1 nearly the entire chain remains consistent with theoretical predictions (with the exception of several frames in the C-terminal section of the chain). The OORF profile visualize opposite role of certain fragments of the chain. In 2PIJ the central fragment (20- 30 windows) represent local maximum, while analogical fragment in 3BD1 reaches its lowest level of RD values. In 2PIJ the RD parameter reaches higher values, especially in the central and the C-terminal fragment of the chain. Both distributions are characterized by low values for the N-terminal fragment (positions 1-10) where the RD parameter does not generally exceed 0.5.
The presence of a complexation partner (marked “P” in Table 3) or a ligand (“L”) does not seem to affect hydrophobicity density distribution in the relevant areas. In general, whenever ligand interaction requires a large discordant cavity, the corresponding deviation can usually be noted by deficiency of hydrophobicity density which can be identifying on the distribution profile (which is not the case here) [32]. Similarly, protein complexation often occurs in areas of excess hydrophobicity exposed on the protein surface – which, again, is not the case with the presented protein [33].
By comparing the results presented in Table 3 and Figure 6, we can conclude that 3BD1 possesses a more stable structure, resembling the idealized “fuzzy oil drop” (i.e., with limited differences between the observed and theoretical hydrophobicity density distributions). Figure 7 reveals variations in the status of each fold.

Figure 6: Distribution of RD values calculated using the OORF system (10 aa Overlapped Open Reading Frame) for two homologous proteins: (A) 2PIJ and (B) 3BD1. Red and yellow fragments denote locations within the sequence of α-helices and β-sheets, respectively.

Figure 7: 3D structure of homologous proteins. A) 2PIJ, B) 3BD1.
Colors indicate the status of each fragment: red areas diverge from the model while cyan ones remain consistent, as shown in Table 3.
Unrelated proteins of common fold
Two proteins: 3CHY wild-type CheY from Escherichia coli, where residue Asp-57 (supported by Lys-109) undergoes phosphorylation and 1RCF - oxidized recombinant flavodoxin from the cyanobacterium Anabaena 7120 responsible for electron transfer from photosystem I to ferredoxin-NADP(+) reductase [22].
The distributions of expected and observed hydrophobicity density distribution in both proteins reveal the high similarity between these two profiles. It is also expressed by low values of RD: 0.300 (O|T=0.089, O|R=0.207) for 1RCF and RD=0.443 (O|T=1.147, O|R=0.185) for 3CHY. However the secondary fragments representing the status of RD>0.5 were found. The helical fragment (112-128) in 3CHY appeared to represent the status discordant versus the model as well as the loop (76-81). Two α-structural fragments (48-54, 120-122) in 1RCF represent the status discordant versus the model as well as the loop 90- 98 (Figure 8).

Figure 8: Profiles of theoretical (blue) hydrophobicity, observed (red) hydrophobicity and RD (black) values in 10 aa OORF system distributions: (A and B) 1RCF, (C and D) 3CHY. Green shades between/under the profiles indicate local hydrophobicity discrepancies. Meaning of red and yellow fragments is the same as in Figures 2 and 6.
The location of fragments representing the distribution of hydrophobicity density in 3D structure of both proteins appeared to be different. The fragments placed rather on the surface of protein in 3CHY represent the discordant status while in 1RCF the dissimilarity versus the model is occurring in the central part of the molecule (Figures 8A- 8D). This observation may suggest different instability of these two molecules, assuming that other than regular ordered hydrophobicity density distribution may influence the local stability. The biological activity of 3CHY requires complexation with other protein molecule [21]. The discordance identified on the protein surface suggests potential area ready for complexation as may be concluded from the FOD model.
Local instability (local discordance observation versus β-expectation) in this case implies substantially different potential tendency to structural differentiation. The lower stability may be supposed in 1RCF as the disagreement is localized in the core of the molecule makes the structural less stable in comparison to analogical β-structural part in 3CHY (Figure 9). It may suggest the easier destabilization of entire molecule (1RCF) while the stable core in 3CHY may protect the molecule against decomposition of the central part of the molecule.

Figure 9: 3D presentation of (A) CHY and (B) 1RCF.
One shall note that the divergence entropy used to measure the differences between profiles recognizes as different positions of opposite tendency. Even large surface between profiles may be ignored by divergence entropy calculation as long as two profiles represent similar tendency.
Green space-filling-residues engaged in biological activity (according to Volz and Matsumura [21])
Yellow fragments–RD above 0.5 recognized according to OORF calculation.
Red fragments–discordant fragments according to RD calculated for secondary fragments.
These two proteins not related one to the other (sequence similarity of only 19% (Clustal2.1 calculation with standard parameters) shows that the structural similarity does not necessarily ensures similar stability of the protein taken the interpretation of FOD model as the criteria for stability estimation.
The study of sequence-to-structure correlations in proteins has a long history [34]. This work hints at the importance of the hydrophobic core in determining the protein’s tertiary conformation.
Our observations support the suggestions contained in Bakker [35], where the authors conclude that protein structure remains tolerant to residue substitutions as long as the hydropathic profile of the sequence is preserved. Since water is an important factor in this process, much effort has been directed towards analyzing the influence of the proteins’ aqueous environment.
They way in which residue sequences encode 3D structures remain a fundamental question in biology. One approach to understanding the folding process is to design a pair of proteins with maximum sequence identity but with differing folds. Therefore, the nonidentity’s must be responsible for determining which fold topology prevails and constitute a fold-specific folding code. The intentionally designed proteins GA88 and GB88, with 88% sequence identity but different folds and functions [36] are described here in the context of the FOD model. Despite a large number of mutations which together bring sequence identity from 16% to 88%, GA88 and GB88 maintain their distinct wild-type 3-alpha and alpha/beta folds, respectively. As the Alexander et al. [36] claim, the 3D-structure determination of two monomeric proteins with such high sequence identity but different fold topology is unprecedented. The geometries of seven nonidentical residues (of 56 total) provide insight into the structural basis for switching between 3-α and α/β conformations. The FOD model applied to these two de novo designed proteins, to two wild-type proteins with intentionally modified sequences as well as to two homologous proteins, reveals the importance of hydrophobic core structure. Our analysis proves that the hypothesis expressing the dominant role of hydrophobic interactions in tertiary structural stabilization can be confirmed quantitatively. Additionally the role of hydrophobic core in stabilization of structures of natural proteins modified (mutated) to converge to a point of highlevel sequence identity while retaining their respective wild-type tertiary folds, of natural proteins with common ancestry but with differing structures and biological profiles shaped by divergent evolution as well as of natural proteins of high structural similarity with no sequence similarity and different biological function was recognized as main mechanism for structure stabilization as expressed by FOD model.
The FOD model posits a structure which consists of a hydrophobic core (central part of the protein body) together with a sheath acting as a buffer zone between the hydrophobic center and the hydrophilic surface. The role of water in the protein folding process and its influence on the final structure of the protein remains a persistent subject in molecular biology [34,37]; however, the question of generalizing the presented observations remains an open issue. The application of FOD model for amyloidosis mechanism is presented Roterman et al. [29]. The applicability of FOD was tested on few selected sets of proteins of small size [38], structural similarity [39], protein complexes [40] and intrinsically disordered proteins [41].