Drug Designing: Open Access
Open Access
ISSN: 2169-0138
ISSN: 2169-0138
Research Article - (2016) Volume 5, Issue 3
Background: Mutations of Estrogen receptors 1 affect its ligand-binding domain and results in the formation of breast cancer. Breast cancer is a known universal disease and the second leading reason behind the death of most females. About 70% of breast cancers express the estrogen receptor. This study was undertaken to realize perceptions into molecular mechanisms and structural necessities that are crucial for potential inhibition of ESR1.
Methods: In this research ESR1 proteins were selected and pharmacophore models were generated, virtual screening was done to obtain hit compounds against reference shared feature pharmacophore, the hit compounds were docked with ESR1 proteins.
Results: The pharmacophore displayed three main features Hydrogen bond acceptor, Hydrogen bond donor and aromatic rings. 10 hit compounds were obtained by virtual screening; compounds were further sorted for Lipinski rule of five before docking. Compounds that fulfill all properties of Lipinski rule of five were docked with proteins, 3 compounds demonstrated ideal docking results. They fit appropriately in the pocket of proteins which demonstrated the soundness and stability of ligand compounds.
Conclusion: It is suggested that these three compounds can be used in the treatment of ESR1 mutations in breast cancer and novel compounds can be designed on the basis of shared feature pharmacophore model for the treatment of ESR1 mutations in breast cancer.
<Keywords: ESR1; HBD; HBA; Molecular docking; Pharmacophore; Virtual screening
It is a known fact that Breast cancer is a known universal disease, with an annual prevalence of 1.3 million cases each year, accounts for more than 23 diseases among all malignancies Breast cancer is more common. In spite of major advances and treatments in its early detection, breast carcinoma still remains a significant reason for morbidity and mortality of women around the world [1]. Breast cancer is the second leading reason behind the death of most females. The most common measure of the treatment of breast cancer is surgery typically followed by adjuvant radiation, chemotherapy, and endocrine therapy, but in a few cases, only medication is given along with radiotherapy [2]. About 70% of breast cancers express mutations in estrogen receptor (ER), while most of the breast cancers demonstrate sensitivity to the inhibition of ER. However, due to unknown facts and reasons, several tumors become unmanageable to ER inhibition during metastatic breast cancer [3].
Estrogen receptors are the member of the nuclear receptor family, thus tend to act as a ligand-activated transcription factors. The binding of ligand prompts a conformational change in the receptor, which results in its translocation into the nucleus, thus activates transcription of several target genes [4]. Mutations of Esterogen Receptor (ESR1) affect its ligand-binding domain. ESR1 are a key mechanism in accomplishing endocrine resistance in breast carcinoma therapy. The ordinary mutations occur in ER Ligand binding domain results in mutations of Tyrosine/Serionine/Aspargin 537, Asp 538, Glycine, Glutamine, Leucine 536, Methionine 543, Leucine 544 and Aspartic acid 531 amino acid residues [5]. Hormone therapy is usually accustomed to inhibit estrogen receptor signal or block ER production. It is at first effective within the roughly seventy percent of patients with breast carcinoma, who have ER-positive tumors; however, several patients develop resistance after long-term exposure to anti-cancer drugs [6]. Therefore, there’s a great need of novel drugs development to cure mutations of ESR1.
Nowadays Pharmacophore approaches became one of the foremost tools in drug discovery after the past century’s development. Numerous ligand-based and structure-based strategies are developed for improved pharmacophore modeling with success and extensively applied in virtual screening, de novo design and lead improvement [7]. Pharmacophores are used as queries for recovering likely leads from structural databases for designing molecules with specific desired attributes and for evaluating similarity and variety of molecules manipulation pharmacophore fingerprints. It may be used to align molecules based on the 3D arrangement of chemical structures or to improve prognostic 3D quantitative structural activity relationship (QSAR) models [8]. Similarly, Virtual screening is a computational process used in the areas of drug discovery and development to explore libraries of small ligands which can be suitably bound to their target proteins or enzymes while docking is a phenomenon of predicting the orientations of molecules in the bounded stable complex.
.To date, there has little information on 3D-QSAR and pharmacophore studies of ESR1 inhibitors. Herein, we tend to report the appliance of pharmacophore modeling, virtual screening and molecular docking for ESR1 inhibitors. This study was undertaken to realize intuitions into molecular mechanisms and structural necessities crucial for potential inhibition of ESR1. That may be helpful within the design of novel ESR1 inhibitors.
The methodology used in this work is shown in Figure 1.

Figure 1: The method applied to design a pharmacophore model for ESR1 mutations in breast cancer.
Screening and selection of ESR1 proteins
After screening Breast cancer inflicting genes the ESR1 gene was selected from Gene cards database [9]. In research collaboratory for structural Bioinformatics, protein data bank (RCSB PDB) in refinement filters, the organism was selected as Homo Sapiens, taxonomy as Eukaryota only, experimental method was selected as x-ray crystallography with an x-ray resultion of 2.0-2.5 Å, which result in best nine ESR1_HUMAN proteins, among those nine proteins three mutated protein ids with similar structures named as 1UOM, 2JFA and 4XI3 and one wild-type ESR1 protein id named as 1R5K were downloaded. RCSB PDB is basically a database that contains X-ray crystallographic and nuclear magnetic resonant 3D structures of proteins and nucleic acids [10].
Preparation of proteins
1UOM is a 3D structure of ESR1 protein in complex with 2-phenyl-1-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-1,2,3,4-tetrahydroisoquinolin- 6-ol ligand with a molecular formula C28 H32N2O2, 2JFA is a 3D ESR1 protein structure in complex with affinity selected co-repressor peptide raloxifene ligand having molecular formula C28H27NO4S, 4XI3 is a 3D structure of ESR1 ligand binding domain in complex with the bazedoxiphene (1-{4-[2-(azepan-1-yl)ethoxy] benzyl}-2-(4-hydroxyphenyl)-3-methyl-1H-indol-5-ol) ligand with a molecular formula C30H34N2O3 and 1R5K is normal human estrogen receptor ligand binding domain respectively. The 1UOM, 2JFA and 4XI3 proteins along with their ligands and wild-type 1R5K protein is shown in Figure 2. The structures were imported into LigandScout software and the protein preparation wizard was used structural alignment of proteins, to ensure structural correctness of the protein structures with high confidence structures. The aligned protein structure was further reserved for the docking and pharmacophore analysis.

Figure 2: Three-dimensional structure of proteins along with ligands a) 1UOM protein structure along with tetrahydro isochiolin ligand b) 2JFA protein structure along with co-repressor peptide ligand c) 4XI3 protein structure along with bazedo xiphene ligand d) wild-type 1R5K protein structure.
Pharmacophore generation
The pharmacophoric features of every ligand were checked in LigandScout software package and their shared feature pharmacophore was designed. Ligandscout is an associated automatic Pharmacophore Model Creation package [11]. The resulted shared feature pharmacophore contains the functional groups involved in their bioactivity towards targeted proteins.
Virtual screening of hit compounds against shared feature pharmacophore
The shared feature pharmacophore model was exported into an alignment tab of LigandScout and set as a reference; virtual screening was done against shared feature pharmacophore to obtain hit compounds, similar to shared feature pharmacophore in the Zinc database with the assistance of Ligscree Server [12].
Generation of shared feature protein
1UOM, 2JFA, and 4XI3 proteins were aligned together in the alignment tab of LigandScout for structural alignment and their shared feature mutated protein structure was designed.
Validation of Hit compounds on the basis of Lipinski rule of five
The hit compounds obtained were then checked for Lipinski rule of five. Lipinski rule of five states that drug-like compound must have HBD less than 5, HBA less than 10, molecular weight no more than 500 Da and logP ranges between 0-5 [13].
Docking of Hit compounds with ESR1 proteins
The compounds fulfilling Lipinski rule of five were docked with wild-type and mutated shared feature ESR1 protein by the patch dock server. Patch dock is usually an algorithmic program utilized for molecular docking, geared towards finding docking transformations and produce smart molecular shapes [14]. Docking results were analyzed and compared with discovery studio; the pharmacophore models of hit compounds were prepared in LigandScout software.
Wild-type ESR1 protein ligand binding domain consists of total 261 amino acid residue and composed of A, B and C chains. Mutated 1UOM ESR1 protein consists of 254 amino acids and consists of only one chain A. Three mutations: C381S, C417S, C530S were identified in 1UOM protein. Mutated 2JFA ESR1 protein consists of 252 amino acids comprising of A, B and C chains, about 4 mutations were found in 2JFA: M361S, M411S, M483S and M530S. Mutated 4XI3 ESR1 protein consists of 243 amino acid residues comprising of A and B chains. The two mutations found in 4XI3 protein: L372S, and L536S. The wild-type and mutated proteins were selected to compare the effects of pharmacophore models on the normal and mutated proteins.
Pharmacophore analysis is measured as an essential portion of drug design. The pharmacophore generated by LigandScout for the selected proteins data set of breast cancer showed three main features Hydrogen bond acceptor (HBA), Hydrogen bond donor (HBD) and aromatic rings (AR). In each pharmacophore model of selected proteins the red arrows represent Hydrogen bond acceptor, green arrow represents Hydrogen bond donor and yellow spheres represent an aromatic ring. Numerous excluded volumes were also produced in the models to demonstrate the space balancing. All the 3 models contain hydrogen bond acceptors, hydrogen bond donors, and aromatic rings. The representative pharmacophores of 1UOM, 2JFA and 4XI3 protein ligands are shown in Figure 3.

Figure 3: 3a: Pharmacophore model of 1UOM protein-ligand, 3b: Pharmacophore model of 2JFA protein ligand, 3c: Pharmacophore model of 4XI3 protein-ligand.
No ligand was attached to wild-type 1R5K protein. Pharmacophore models of selected protein data sets were aligned together on the basis of structure, to generate a shared feature pharmacophore shown in Figure 4.

Figure 4: Model of shared feature pharmacophore of 1UOM, 2JFA AND 4XI3 protein structures ligands.
In the Virtual screening, 10 hits compounds similar to share feature pharmacophore model were obtained with a Z-score value of 1.72898. The Z-score is used to estimate the quality of either ligand compounds or protein structures as an inhibitor compound; using the structures of already commercially available drugs as well as structures of solved proteins as references [15]. The hit compounds were then checked for Lipinski rule of five only five compounds were fulfilling all the rules of Lipinski, i.e., molecular weight<500 Da, HBD<5, HBA<10 and logP between 0-5. The hit compounds which fulfilled Lipinski rule of five are shown in Table 1.
| Compounds | Molecular formula | Molecular weight | LogP | HBD | HBA |
|---|---|---|---|---|---|
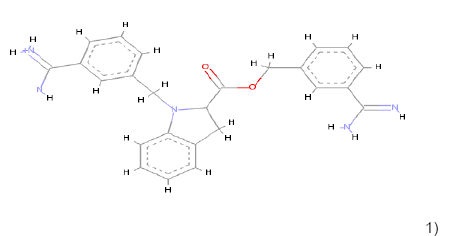 1) 1) |
C25H26N5 O2 | 428.209 | 2.0 | 2 | 5 |
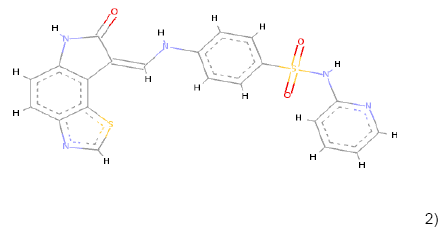 2) 2) |
C21H15N5O3S2 | 449.062 | 1.65 | 1 | 8 |
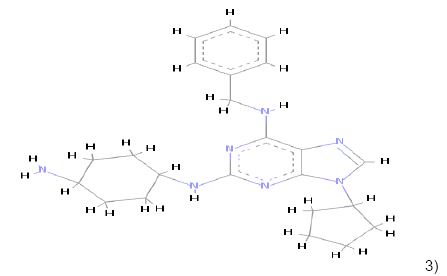 3) 3) |
C23H31N7 | 405.264 | 2.46 | 1 | 6 |
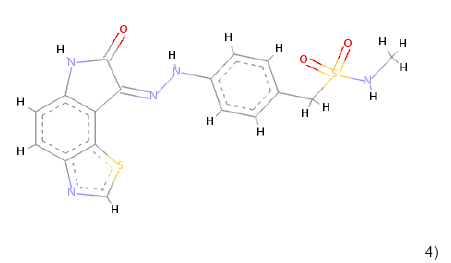 4) 4) |
C17H15N5O3S2 | 401.062 | 1.35 | 1 | 8 |
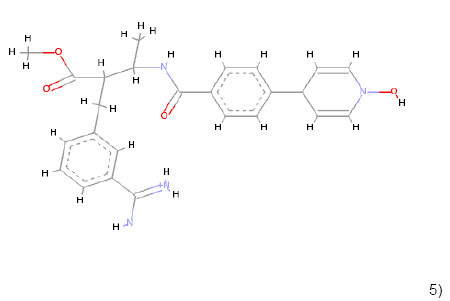 5) 5) |
C25H29N4O4 | 449.219 | 1.93 | 2 | 6 |
Table 1: Chemical structures, molecular formulae, molecular weights, logP, HBD and HBA of hits compounds fulfilling the Lipinski rule of five.
Morris and Lim reported that Molecular docking is a vital tool in the structural molecular biology and the computer-aided drug design. The aim of ligand-protein docking is to foresee the principal binding modes of a ligand with a known 3D structure of protein [16]. All satisfied compounds were docked with mutated shared feature ESR1 protein and wild-type ESR1 protein, in every docked complex the common interacting amino acid residues were same as that of pharmacophore models of 1UOM, 2JFA, and 4XI3 proteins, but among the compounds represented in Table 1, the 1st and 4th compound produced more than five bumps with both proteins after docking. Bumps refer to the collision of molecules to each other; the minimum acceptable number of bumps is five. The 2nd, 3rd and 5th compounds demonstrated ideal docking results with wild type and mutated shared feature ESR1 protein structures. The three compounds with best docking results are shown in Table 2.
| Compounds | Bonds formed in docking with mutated shared feature ESR1 protein | Bonds formed in docking with wild type ESR1 protein |
|---|---|---|
1) 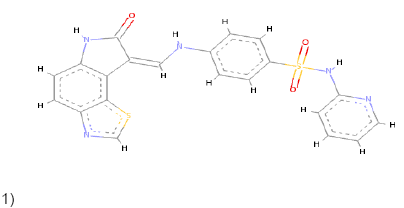 |
Conventional Pi-Pi T shaped Pi-Alkyl Pi-Anion Pi-Sulfur |
Conventional Pi-Pi T shaped Pi-Alkyl Pi-Donor Pi-Sulfur |
2) 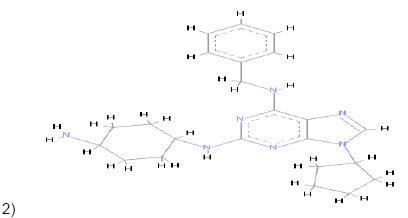 |
Carbon Pi-Donor Pi-Alkyl Pi-Anion Pi-Pi T shaped |
Pi-Pi T shaped Pi-Alkyl Alkyl Carbon Pi-sigma |
3) 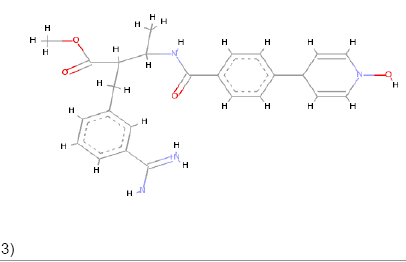 |
Pi-Alkyl Pi-Anion Charge-Charge Pi-Carbon |
Pi-Sulfur Pi-Donor Pi-Sigma Alkyl Amide Pi Stacked Carbon |
Table 2: Comparison of bonding types of compounds which demonstrated ideal docking results along with both wild type and mutated shared feature protein structures.
Compounds represented in Table 2 demonstrated better interactions with interacting amino residues and best fit in the pockets of both proteins. In both the wild-type and mutated protein dockings, similar bondings were observed, whereas, in wild-type docked complex some extra bondings were also observed that determine the stability of hit compounds to better use as drugs. The docking results and interacting amino acid residues for both wild type and mutated shared feature protein are shown Tables 3 and 4.
| a) | 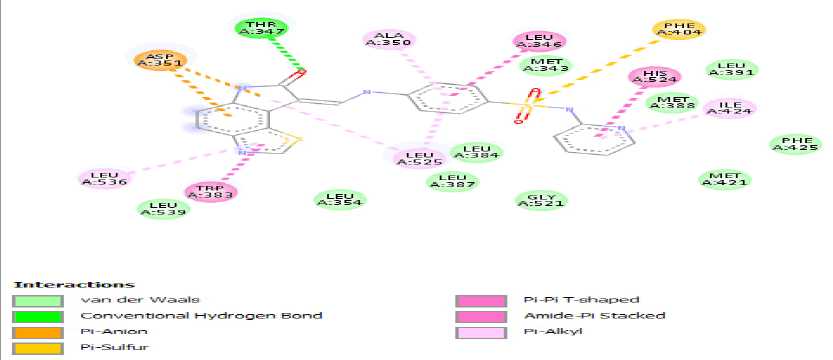 |
| b) | 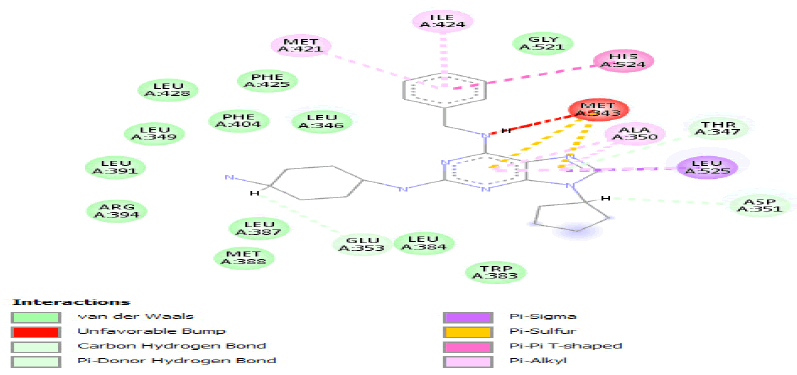 |
| c) | 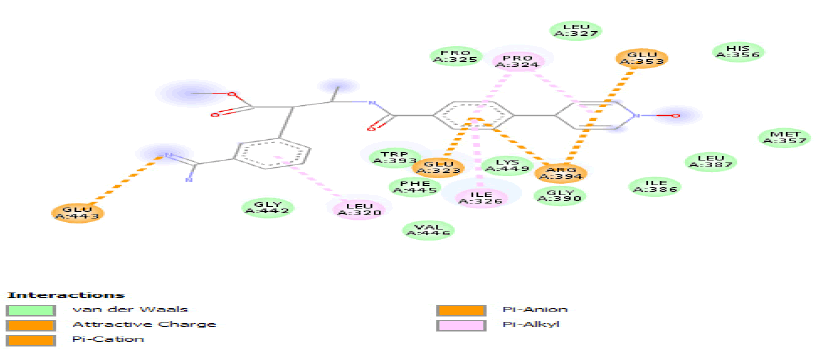 |
Table 3: Ideal docking results of compound 2, 3 and 5 with mutated shared feature ESR1 protein.
| a) | 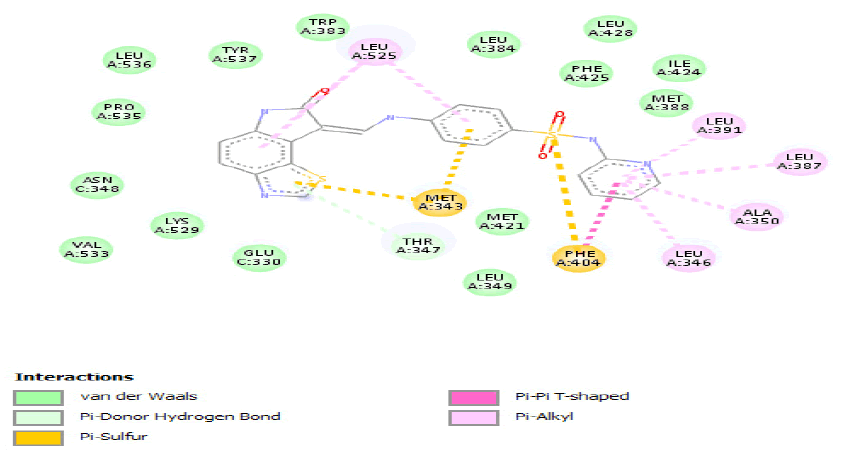 |
| b) | 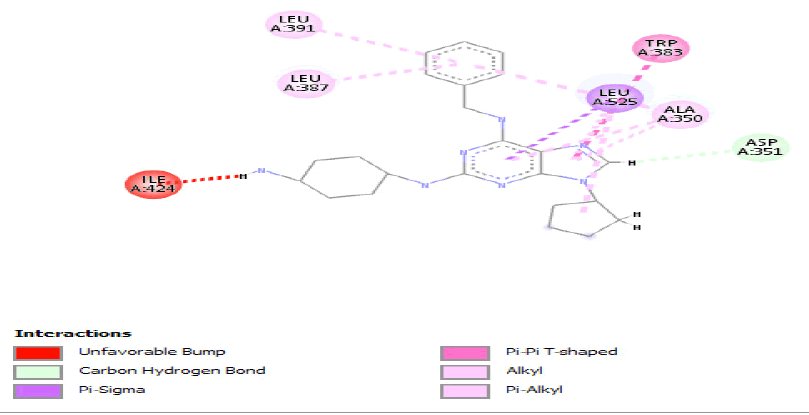 |
| c) | 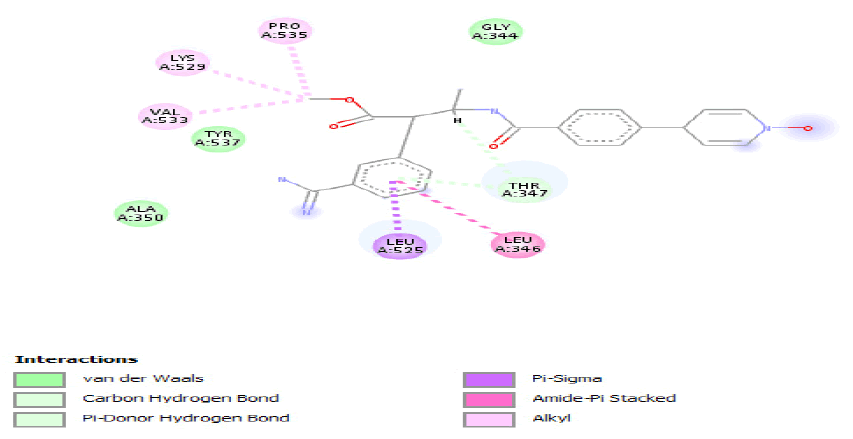 |
Table 4: ideal docking results of compound 2, 3 and 5 with wild type ESR1 protein.
The pharmacophore models of three superlative hit compounds docked with protein structures were generated in LigandScout to get information about HBD, HBA and ARs. The pharmacophore models of these compounds are shown in Figure 5.

Figure 5: Pharmacophore models of compounds demonstrated ideal docking results with ESR1 proteins.
All these three compounds are best suitable to use as drugs as they fulfill all the properties of Lipinski’s rule so they will demonstrate fewer side effects as compared to the drugs available in the market. It is suggested that these three compounds can be used in the treatment of ESR1 gene mutations in breast cancer.
The pharmacophore model is a very convenient tool for new lead compounds detection and development. Pharmacophore development is the first step towards understanding the collaboration between a receptor and a ligand. It was often suggested as the “spirit” of the structure-activity information extended to date. A pharmacophore model is a sensible qualitative prediction of binding by identifying the three-dimensional arrangement of small number atoms belongs to functional groups [17]. Two common approaches used in pharmacophore modeling are ligand based and structured based. Ligand-based pharmacophore modeling adapts the superposition of a set of active compounds and extracting shared chemical features essential for the bioactivity of molecule whiles structure based pharmacophore modeling adapts the mechanism of examining promising interactions between receptor and ligand [18].
In this research work structure based pharmacophore modeling approach was utilized. The combinations of the pharmacophore, virtual screening, and molecular docking positively give possible inhibitors that can have boundless influence for various experimental studies in diseases [19]. The chemical features of compounds discovered by the interaction are taken into interpretation into the structure-based models, as well as the interactions between the target receptor and the ligand molecule [20]. Virtual screening of ligand libraries results in the important approaches for drug discovery in the drug discovery process virtual screening of drug database is an alternative attempt to high throughput screening techniques. Drug-likeness properties of compounds are important strategies in selecting drug compounds that satisfy the Lipinski rule of five [21].
Z score value was used to obtain hit compounds, usually Z score is a measure of the fit of the ligand to the active pocket of receptors, it also uses to predict the binding affinity of molecules. The hit compounds obtained in the study were further sorted for Lipinski rule of five to obtain lead compound for drug discovery against breast cancer. Finally; five compounds were selected for docking. The molecular docking approach was used to further confirm the hit compounds as inhibitors for the ESR1, molecular docking predicts the structures of the intermolecular complexes formed between the two or more molecules [22,23]. Among the docked results 1st and the 4th compound produced more than five bumps with protein, so they were discarded because the favorable number of allowed bumps is less than five. In docked result ligand and interacting residues must be far apart from each other for proper interaction. Though, when they become very near, they bounce off of each other tremendously fast, one molecule smashes into another hence they destroy themselves [24]. Structural alignments of proteins were done to produce a shared feature protein structure. Structural alignment is thought to be as an important and critical step in pharmacophore analyzes because this affects the reliability of the models. The structure-based pharmacophore model can be derived straight from ligand-protein co-crystallized structure and therefore, can imitate more consistent combination of the critical features obligatory for relating biological potency [25].
The structure-based pharmacophore and virtual screening results help to foresee the biological actions of the compounds with modification in the chemical substitutions and offers useful orientations for the design of novel inhibitor compounds [26]. Structure-based pharmacophore model utilizes the interactions between receptor-ligand complexes to generate a hypothesis. An originating pharmacophore model for the 3-D structure of a target protein offers useful information for investigating protein-ligand interactions and further development of ligand binding attraction. Though, pharmacophore model derived from previously known inhibitors enables the identification of vital chemical features existing in experimentally known potent inhibitors [27]. Although Tamoxifen drug is available in the market for the treatment of estrogen receptor mutations in breast cancer as it directly bound to the ESR1 gene, but Tamoxifen is linked to increased chances of endodermal and uterus tumors. Unluckily, advanced breast cancers become unmanageable to Tamoxifen treatment, their heterotrophic action also limits its use [28]. These pharmacophore models might be useful to consider the inhibitory actions; and in future explorations of novel drug compounds in breast cancer.
The present work was done to find novel inhibitors of ESR1 by the in silico method, which specifically binds to its active site. Understanding of the key basic structural characteristics for ESR1 inhibition has been accomplished by applying structure-based pharmacophore model configuration, virtual screening, and molecular docking strategies. The utilization of 3D structures of mutated ESR1 proteins has uncovered a pharmacophore model showing the key features essential for inhibitor binding. The produced pharmacophore contains three primary features; HBD, HBA and AR. In future, these pharmacophore models will assist to discover new antitumor compounds, obtained potential inhibitors can be purchased and tested in vitro against the ESR1 and different cancer cell lines, to test its adequacy and social advantage. Further experimental methods can be conducted to determine the efficacy of these pharmacophore models as inhibitor compounds.
Authors seriously acknowledge the sustenance delivered by the Department of Bioinformatics and Bioinformatics research club Government Post Graduate College Mandian for conducting this research work. The authors also thanks to Mr. Muhammad Haseeb from Capital University of Sciences and Technology, who provided beneficial information to conduct research work.
This research work is unique and has not been submitted to any other journal. None of the authors have challenged conflicts of interest.
Anum Munir carried out entire research process, gathers data, analyzed and interprets the results and also wrote Research paper under the supervision of Shumaila Azam and co-supervision of Sahar Fazal. Azhar Mehmood, Arshad Mehmood and Zanib khan help in the analysis of pharmacophore models and docking results. The manuscript final draft proofreads and approved by all of the authors.