Pancreatic Disorders & Therapy
Open Access
ISSN: 2165-7092
ISSN: 2165-7092
Review Article - (2014) Volume 4, Issue 3
Hypertriglyceridemia has been estimated to cause around 7% to 10% of the total number of cases of acute pancreatitis in man. The mechanisms involved in the progression of hypertriglyceridemia to pancreatitis are not well understood.
In this paper, we refer to different mechanisms proposed by previous authors to explain the pathogenesis. We discuss these theories in relation to our own experimental results. It has been difficult to verify previous theories because pancreas biopsies cannot easily be obtained in humans in the early, mild stages of acute pancreatitis. In patients with severe pancreatitis, the pancreas is typically grossly pathological. We have used an animal model with lipoproteinlipase deficiency. In these animals, severe hypertriglyceridemia can be induced by feeding a high-fat diet, which in turn induces the development of severe pancreatitis. By sacrificing animals at different times, it was possible with the use of light- and electron microscopy to monitor the development of acute pancreatitis from the earliest detectable changes to the most advanced, full-blown stages.
We found that the earliest detactable changes consisted of a selective degeneration of the mitochondria in the exocrine cells. At the same early stage the other intracellular structures, including the endoplasmatic reticulum, were well preserved. Mitochondria are known to be the major source of cellular free radicals liberated by the electron transport chain which may lead to oxidative stress. In normal cells antioxidant mechanisms such as vitamin E, glutathion peroxidase and others will neutralize free radicals and thus prevent oxidative stress. Our finding of an initial mitochondrial degeneration, probably caused by free fatty acids deranging the mitochondial function, support the view that antioxidants may have a role in the prevention of recurrent hypertriglyceridemia-induced pancreatitis.
Keywords: Acute pancreatitis, Chronic pancreatitis, Hypertriglyceridemia, Hyperlipemia
FFA: Free Fatty Acids
The detailed pathophysiological mechanisms involved in the progression of hypertriglyceridemia to pancreatitis are little known and little studied. In this paper, we refer to different mechanisms proposed to explain the pathogenesis by previous authors and discuss these theories in relation to our own experimental results. We used an animal model with lipoproteinlipase deficiency to study this progression. In these animals, severe hypertriglyceridemia can be induced by feeding a high-fat diet, which in turn induces the development of severe pancreatitis. By sacrificing animals at different times, it was possible with the use of light- and electron microscopy to monitor the development of acute pancreatitis from the earliest detectable changes to the most advanced, full-blown stages.
In man, acute pancreatitis in an initial mild stage is not an indication for pancreatic surgery, meaning that biopsies cannot easily be obtained [1]. In patients with severe acute pancreatitis, the pancreas is typically grossly pathological and in fatal cases the gland is changed by rapid autolysis and postmortem changes. Animal models such as ours therefore provide the only useful way of studying the development of the disease. At the moment, our mink model with lipoprotein lipase deficiency appears to be the only known animal model in which hypertriglyceridemia causes pancreatitis.
Hypertriglyceridemia has been estimated to cause around 7% to 10% of the total number of cases of acute pancreatitis in man [2]. Gall stones and alcoholism are other well-known causes. Pancreatitis is also known to be associated with conditions involving poorly regulated fat metabolism, such as diabetes and obesity [2]. A delectable dinner with a combination of alcohol and delicious, fatty food can provoke a severe hypertriglyceridemia in predisposed persons.
The diagnosis can easily be missed. A hypertriglyceridemia present in the initial stage of pancreatitis, can rapidly subside in patients with acute abdominal pain. They lose appetite, and after admission to a hospital a fasting state is prescribed. Blood triglyceride concentration should therefore be measured as early as possible if one is to reveal hypertriglyceridemia as the cause of the disease.
Previous theories proposed to explain the pathogenesis
Several theories have been proposed to explain the pathogenesis of pancreatitis in hypertriglyceridemia.
1. A commonly accepted hypothesis was suggested by Havel as early as 1969 [3]. In short, excessive amounts of circulating triglyceride-rich lipoprotein could be hydrolyzed by inappropriately-activated pancreatic lipase. Free fatty acids (FFA) could then be liberated in toxic concentrations, causing cell damage.
2. Another theory favors plasma hyperviscosity due to elevated concentrations of chylomicrons as the underlying pathophysiological principle. The hyperviscosity is reputed to lead to ischemia and acidosis in pancreatic capillaries [4,5].
3. Other theories focus on a role of oxidative stress in the pathogenesis of hypertriglyceridemia-induced pancreatitis. The suggestion is that free radicals, which are a normal product of cellular metabolism, are produced in quantities exceeding the capacity of radical-scavenging mechanisms to absorb them. Contributing factors could include a relative deficiency of vitamin E, selenium or other unknown antioxidants [6-9].
Recent results using an unusual animal model: We used mink with lipoprotein lipase deficiency to study the mechanism of the pancreatitis. Blood samples were routinely drawn from juvenile farmed mink to diagnose a serious, infectious plasmacytosis. During the course of this investigation, it was accidently discovered that some individuals had severe hypertriglyceridemia [10,11]. Feed of high fat content is often used on mink farms to obtain high fur quality at reasonable feed cost. It was found that affected individuals were homozygotic for lipoprotein lipase deficiency and the defect in the lipoprotein lipase gene was characterized [12]. This seems to be the only known animal model in which pancreatitis can be induced by hypertriglyceridemia. Another animal model in cats with lipoprotein lipase deficiency has been extensively studied, but these cats do not develop pancreatitis [13].
By systematically breeding heterozygotes, we were able to obtain a sufficient number of homozygotes for further studies. As soon as the pups were weaned, blood samples could be drawn, homozygotes could be identified and their fat metabolism could be studied. By sacrificing animals at different time points relative to the development of pancreatitis, the gross anatomy, light microscopy and electron microscopy of both normal and progressively more damaged pancreases could be studied. All homozygotic animals tested developed pancreatitis at an early age. Later, fatty liver and splenomegaly were common findings.
The earliest detectable changes in the pancreas occurred in the mitochondria of exocrine cells. Light microscopy showed multiple cytoplasmatic vacuoles in exocrine cells in pancreatic acini (Figure 1b) as compared to normal cells (Figure 1a)
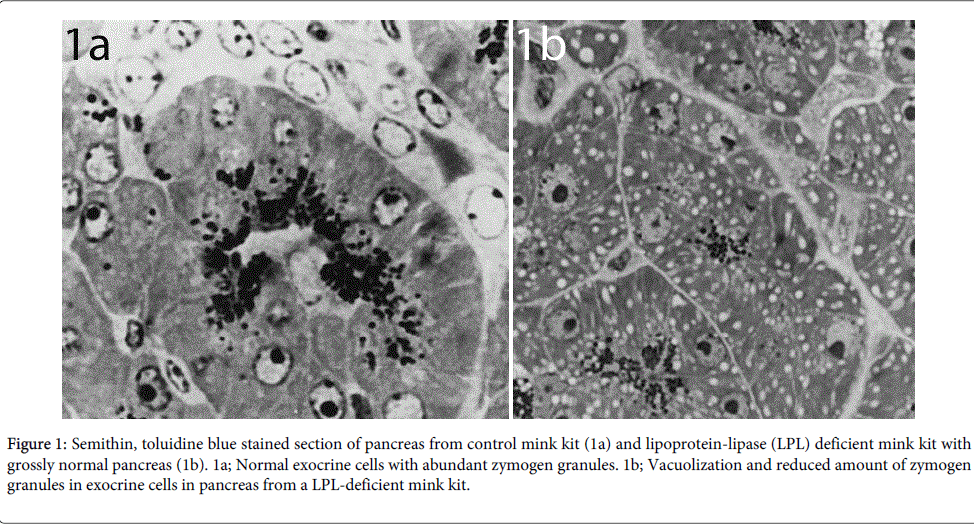
Figure 1: Semithin, toluidine blue stained section of pancreas from control mink kit (1a) and lipoprotein-lipase (LPL) deficient mink kit with grossly normal pancreas (1b). 1a; Normal exocrine cells with abundant zymogen granules. 1b; Vacuolization and reduced amount of zymogen granules in exocrine cells in pancreas from a LPL-deficient mink kit.
This was followed by multicellular necrosis and loss of exocrine cells (Figure 2)
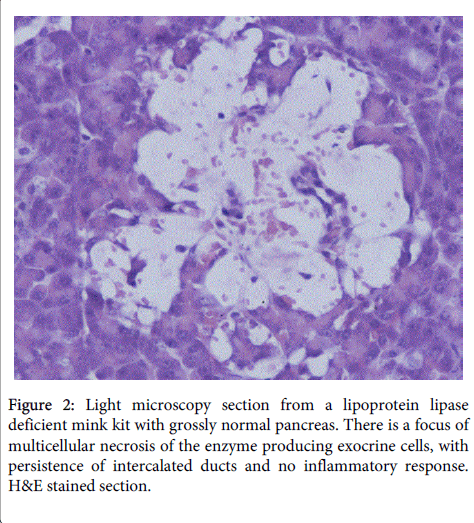
Figure 2: Light microscopy section from a lipoprotein lipase deficient mink kit with grossly normal pancreas. There is a focus of multicellular necrosis of the enzyme producing exocrine cells, with persistence of intercalated ducts and no inflammatory response. H&E stained section.
Electron microscopy showed that the cytoplasmatic vacuolation was caused by swelling of mitochondria with loss of crista (Figure 3).
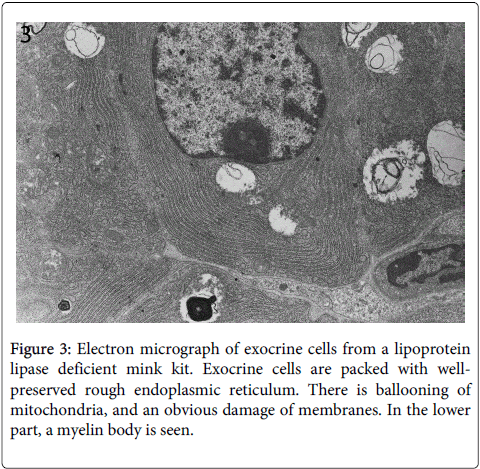
Figure 3: Electron micrograph of exocrine cells from a lipoprotein lipase deficient mink kit. Exocrine cells are packed with wellpreserved rough endoplasmic reticulum. There is ballooning of mitochondria, and an obvious damage of membranes. In the lower part, a myelin body is seen.
In some cases the destroyed mitochondrial membranes formed lamellar myelin-like bodies (Figure 4).
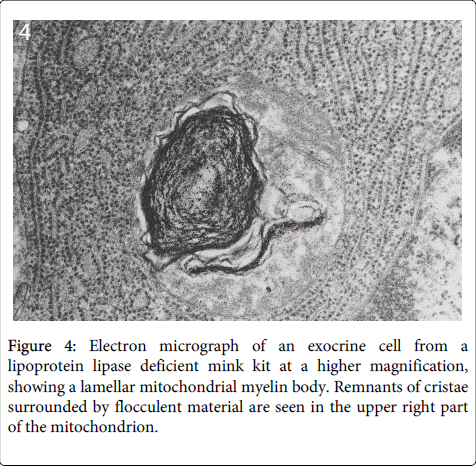
Figure 4: Electron micrograph of an exocrine cell from a lipoprotein lipase deficient mink kit at a higher magnification, showing a lamellar mitochondrial myelin body. Remnants of cristae surrounded by flocculent material are seen in the upper right part of the mitochondrion.
In contrast, the other cellular structures were well preserved at this early stage of cellular destruction, most notably the rough endoplasmatic reticulum (Figures 3 and 4). Also indicating that the cellular destruction starts inside the cells is the finding that intact desmosomes were present (Figure 5).
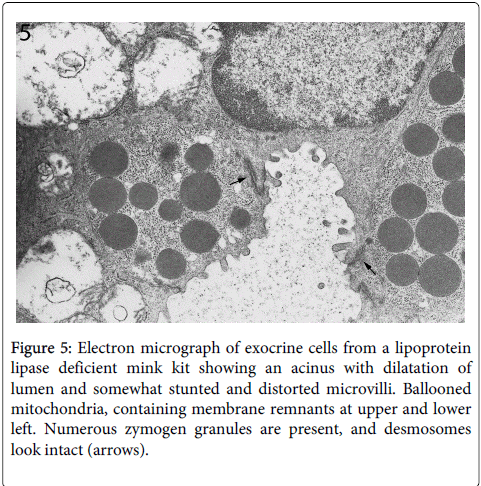
Figure 5: Electron micrograph of exocrine cells from a lipoprotein lipase deficient mink kit showing an acinus with dilatation of lumen and somewhat stunted and distorted microvilli. Ballooned mitochondria, containing membrane remnants at upper and lower left. Numerous zymogen granules are present, and desmosomes look intact (arrows).
Desmosomes are structures that anchor the outer cell membranes of neighboring cells to each other. Results of light- and electron microscopy with the focus on veterinary pathology have been published by us in the Journal of Comparative Pathology [14]. As well known, the rough endoplasmic reticulum, with its abundant ribosomes, is a prominent structure of the exocrine cells because of the synthesis of digestive protein enzymes such as lipase, trypsin, amylase and many others. The protein synthesis in the exocrine pancreas is very active due to its production of digestive proenzymes.
Why should the cellular damage start in the mitochondria?
Mitochondria are known to be the major source of cellular free radicals liberated by the electron transport chain. An excess of FFA in the cells, exceeding the capacity of intracellular fatty acid binding-proteins, can derange mitochondrial function. FFA can uncouple the respiratory electron transport chain and thus liberate free radicals [15]. The hydrophobic end of FFA can penetrate into the lipid double membranes and disturb the membrane structure required for the respiratory electron transport chain [16].
How does our finding of an initial, selective degeneration of the mitochondria compare with previous theories of the pathogenesis of hypertriglyceridemic pancreatitis? According to Havel, pancreatic lipase can be released into the vascular bed of the pancreas, and hydrolyze triglycerides in triglyceride-rich lipoproteins when these are present in excessive amounts [3]. The very high concentrations of FFA thus formed will exceed the binding capacity of plasma albumin. FFA will self-aggregate, forming micellar structures with detergent properties. These FFA micelles will attack platelets, the vascular endothelium and, finally, acinar cells, producing ischemia and pancreatic injury. The resultant ischemia then creates an acidic environment, which further enhances FFA toxicity.
We, however, found no signs of endothelial injury, platelet aggregation or ischemia in the earliest stages, as proposed by Havel [3]. Instead we found an extensive initial mitochondrial degeneration.
Both Havel's explanation and our findings of an initial mitochondrial degeneration are, however, in agreement with the view that an excessive concentration of FFA plays a key role on the pathogenesis of pancreatitis induced by hypertriglyceridemia. Two different sources of the excess of FFA can however be discussed
1. Triglyceriderich lipoproteins in the vascular bed could be the source after hydrolysis by pancreatic lipase
2. Lipoproteins internalized by receptor mediated uptake in the exocrine pancreas cells could be the source.
As proposed by Havel, it is possible that these FFAs could be liberated by pancreatic lipase acting on triglyceride-rich lipoproteins in the vascular bed of the gland. This would require two conditions to be fulfilled. It would require that the lipase proenzyme produced by the exocrine pancreas cells can be activated in situ in the gland itself or in its vascular bed. This activation of the enzyme is traditionally considered to take place in the duodenum. It would also require that VLDL and/or chylomicron can serve as substrates for pancreas lipase. Although this seems to be likely, it has not been directly shown experimentally.
If the selective mitochondrial degeneration is caused by FFA liberated in the pancreatic vascular bed, it would imply that 1) FFA are formed in excess of the binding capacity of albumin and 2) after uptake in the acinar cells the FFA concentration should also exceed the binding capacity of intracellular fatty acid binding proteins [14]. This concentration of FFA could then be toxic to the mitochondria while at the same time not causing visible damage to the external cell membrane or other intracellular membranes such as the endoplasmic reticulum. This may be the case if the mitochondria with their electron transport chain are more sensitive to an excess of FFA than are other lipid membranes such as the vascular endothelium, platelets and the external cell membrane of acinar cells and other intracellular membranes.
As an alternative to Havel's hypothesis, it can be hypothesized that an excess of FFA can be liberated by intracellular hydrolysis of triglyceride-rich lipoproteins after uptake in the acinar pancreas cells [11]. It is well known that chylomicron and VLDL can be taken up as intact particles by cells in most tissues when the concentration of triglyceride-rich lipoproteins is very high. We found that also in lipoprotein lipase-deficient animals, the hypertriglyceridemia subsides on fasting or on a low-fat diet [10]. As is well known, hepatocytes have an abundance of such lipoprotein receptors. Since the pancreas and the liver both originate from the same structures during embryogenesis, it may also be postulated that the pancreas is rich in receptors for triglyceride-rich lipoproteins.
Hyperviscosity: As stated it has been proposed that hypertriglyceridemia could cause hyperviscosity in the pancreatic vascular bed and thus cause cellular ischemia [4,5]. Our finding of an initial mitochondrial degeneration does not support the theory that hyperviscosity plays an important role in the pathogenesis of this type of pancreatitis.
Oxidative stress: As mentioned above, oxidative stress has been proposed to play a role in inducing pancreatitis in hypertriglyceridemia [6]. The causes of the imbalance between free radicals and scavenging mechanisms in hypertriglyceridemia have not been established. Our finding of an initial mitochondrial degeneration agrees with the view that oxidative stress is important in the pathogenesis. In our view it is an excess of FFA that leads to oxidative stress in the mitochondria.
Prevention and treatment
To prevent or treat hypertriglyceridemia is the primary goal in patients suffering from this type of pancreatitis. In hypertriglyceridemia caused by high levels of VLDL and of chylomicron (Fredrichson's type V), dietary change, eicosapentaenoic acid supplementation and specific drug therapy can all be beneficial. In the rare, congenital errors with hyperchylomicronemia (Fredrichson's type I), treatment strategies are more demanding, and may include plasmapheresis.
Antioxidants such as vitamin E have been tried in a few patients in the acute stage of pancreatitis, although the low number of patients included has not provided conclusive results [17]. In one study three patients with frequent episodes of pancreatitis due to hyperchylomicronemia caused by lipoprotein lipase deficiency were treated with high doses of antioxidants (selenium, carotene and vitamin E) [6]. After this regime was started, the number and severity of episodes of pancreatitis were markedly reduced.
Our findings of an initial mitochondrial degeneration, most probably caused by free radicals leading to oxidative stress, support the view that antioxidants may have a role in the prevention of recurrent hypertriglyceridemia-induced pancreatitis. In the future, clinical trials with and without high doses of antioxidants should be carried out in patients suffering from recurrent hypertriglyceridemic pancreatitis.