Journal of Chromatography & Separation Techniques
Open Access
ISSN: 2157-7064
![]() +44 1300 500008
+44 1300 500008
ISSN: 2157-7064
![]() +44 1300 500008
+44 1300 500008
Research Article - (2015) Volume 6, Issue 6
Here we present a communication about the article “Salvatierra-Stamp VC, Ceballos-Magaña SG, Gonzalez J, Ibarra-Galván V, Muñiz-Valencia R (2015) Analytical method development for the determination of emerging contaminants in water using supercritical-fluid chromatography coupled with diode-array detection. Analytical and Bioanalytical Chemistry 407:4219-4226”. In this paper, a selective, linear, accurate and precise supercritical-fluid chromatography coupled with diode-array detection method was developed and validated for the determination of seven emerging contaminants: two pharmaceuticals, three endocrine disruptors, one bactericide and one pesticide. The compounds were base-line separated in around 10 minutes. Also, the method involved a sample treatment optimization by means of C18-OH solid phase extraction cartridges. The developed method was validated. In this sense, the correlation coefficient and recovery was higher than 0.9997 and 94%, respectively. Limit of detection and quantification was in the range of 0.10-1.59 μg/L and 0.31-4.83 μg/L, respectively. The measurement uncertainty was evaluated using the top-down model considering six sources of uncertainty. For all compounds, the uncertainty associated with accuracy and linearity regression was the main contribution to the combined uncertainty. Expanded uncertainties for each compound in method analysis were lower than 10.8%. Finally the method was successfully applied to environmental water samples.
Keywords: Emerging contaminants; Supercritical-fluid chromatography; Solid phase extraction; Water samples; Uncertainty assessment
Emerging contaminants (ECs) consist of a large and growing group of compounds of natural and anthropogenic origin, among which include pharmaceuticals, pesticides, personal care products, hormones, industrial chemicals, etc. [1]. The occurrence of emerging contaminants in the aquatic environment has become an environmental problem of global concern. Traditionally these organic compounds were not considered pollutants; however, today it is known that many of them have potential harmful effects in various organisms, causing toxic effects and disorders in the endocrine system and can cause irreversible effects [2].
Pharmaceuticals and daily personal care products (PCPs) are widely used all over the world; however their disposal and bodyexcretion are usually not controlled [3,4]. A similar situation occurs with household pesticides which are often used without heed and disposed of as household waste. These actions lead to the contamination of the environment with different classes of pesticides [5]. The presence of these pollutants in the environment is usually in a very low concentration range: from ng/L to μg/L.
Sometimes the analysis of these pollutants can be difficult due to the wide variety of compounds. For this reason various techniques are used for both cleaning up and pre-concentration. The most widely used sample preparation is the solid phase extraction (SPE) [4,6,7], while liquid chromatography coupled with mass spectrometry (LC-MS) is the preferred analytical technique due to its high selectivity and sensitivity [8,9]; however, this MS detector requires high operating costs and is not available in many laboratories.
The use of supercritical-fluid chromatography (SFC) is an alternative technique that improves the analysis of these compounds, which has stimulated the interest of many researchers. In this technique, a mixture of carbon dioxide (CO2) and an organic solvent as mobile phase is commonly used, allowing the use of high flow-rates with low pressure falls through the column, leading short analysis time and decreasing the consumption of the organic solvent, making the use of SFC a faster and attractive technique. Despite these advantages there are few publications related to the determination of contaminants in environmental samples [10]. Furthermore there are much fewer publications employing SFC coupled with diode-array detector (DAD) for analyzing emerging contaminants in water samples [11].
Nowadays, the demonstration of reliability and quality of the data produced in chemical analysis is of great importance during method development, especially for accredited laboratories. To ensure the reliability of the results both traceability and estimation of measurement uncertainty are important. This reliability and comparability of the data are obtained from the method validation and uncertainty assessment. The uncertainty is a quantitative indicator associated with a level of confidence, in which the different possible sources of uncertainty are evaluated, including the stock solution preparation, sample preparation, glassware, etc. The aim of this evaluation is to show critical stages of an analytical method where uncertainty could be reduced.
The uncertainty can be estimated using different models being "top-down" and "bottom-up" the most commonly used. The "bottomup" model includes a decomposition process of all unit operations performed by the analyst, grouped into common activities and an estimate of their contribution to the combined uncertainty value of the measurement process. The "top-down" model is based on method validation and precision data derived from the results obtained in the laboratory [12]. This model evaluates each individual uncertainty for every single step.
The EURACHEM/CITAT Guide Quantifying Uncertainty in Analytical Measurement recommends the following steps for proper estimation of uncertainty: i) specify measurand, ii) identify uncertainty sources, iii) quantify uncertainty components and iv) calculate combined and expanded uncertainty. In general, there are few papers about CEs analysis in which the uncertainty is estimated.
The objective of the work reported in the article Analytical and Bioanalytical Chemistry (2015) 407:4219-4226 was to develop, validate and apply an easy and sensible SPE-SFC-DAD analytical method to quantify glyburide, carbamazepine, 17 α-ethinyl estradiol, 17 β-estradiol, bisphenol A, diuron and triclosan in water samples. Moreover to determine the critical stages of the proposed method an estimation of the uncertainty applying the top-down model was carried out. Finally, the combined and expanded uncertainty for each compound was calculated from the contribution of each stage.
Solvents and materials
All analytical standards: carbamazepine (CBZ), glyburide (GBD), 17 α-ethinyl estradiol (17EE), 17 β-estradiol (17E), bisphenol A (BPA), triclosan (TCS) and diuron (DIU) and LC-MS grade solvents (methanol (MeOH) and acetonitrile (AcN)) were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Working standard solutions were prepared weekly, stored at 4°C and protected from light.
The cartridges used for solid phase extraction (SPE) were Bond Elut-C18OH (1 g per 6 mL) and HF Bond Elut-C18 (500 mg per 3 mL) from Varian, Agilent (Santa Clara, CA. USA) and Discovery DSC18 (500 mg per 6 mL) from Sigma-Aldrich (Saint Louis, MO, USA). A Visiprep SPE vacuum manifold from Sigma-Aldrich (Saint Louis, MO, USA) and a vacuum pump model EV-40, from EVAR (Guadalajara, Mexico) were also used.
Collection and treatment of water samples
Different water samples (WS) were collected in amber glass bottles previously rinsed with ultra-pure water at a depth of 0.5 to 1 meter, in rivers near Colima City, Mexico. These samples were kept in coolers and stored at 4°C, protecting them from daylight until analysis. All samples were filtered through 0.45 μm highly hydrophilic polyvinylidene fluoride membrane filters from Phenomenex (Torrance, CA, USA) to eliminate suspended matter.
The SPE protocol used Bond Elut-C18OH cartridges previously raised with 3 mL of methanol and 6 mL of ultrapure water. Then, 150 mL water sample was placed in a 250 mL flask and processed at 8 mL/min flow-rate at pH 5.5 through the cartridge. Subsequently, the elution was performed using 3 mL MeOH at 1 mL/min flow-rate and injected into the SFC-DAD system.
SFC-DAD analysis
Chromatographic separation was carried out on an Acquity Ultra Performance Convergence Chromatography (UPC2) system equipped with a binary solvent pump, with refrigerated auto sampler with a 10 μL loop, back-pressure regulator (BPR), convergence manager and column oven. This equipment was coupled with a diode-array detector (DAD). The BPR was maintained at 2000 psi. The column Viridis BEH-2-EP (4.6 mm × 100 mm, 5 μm particle size) with 10 μL injection loop was used at 40°C. The temperature of the auto sampler was maintained at 15°C. Data collection and analyses were performed using EmpowerTM Pro 3 Software. All system components are from Waters (Milford, MA, USA). Pressurized CO2 (99.999%) was purchased from Praxair (Colima, Mexico).
The separation used gradient elution mode at a flow-rate of 1.4 mL/ min. The gradient elution was performed as follows: from 0 to 5 min increase from 5 to 30% AcN; and finally from 5 to 10 min increase from 30 to 40%. Column reequilibration was performed for 3 min at the initial conditions. The qualitative analysis was performed in the 190-360 nm range, using the UV-absorbance DAD detector. Quantitative analysis was performed at 215 nm. ECs peak identification and the evaluation of purity each EC peak was done by comparing their retention times and UV spectra.
Method validation
Method validation was performed following the ICH and European Commission Decision 2002/657/EC guidelines [13,14], assessing selectivity, linearity, precision, accuracy and limits (quantification and detection).
Measurement uncertainty
The uncertainty was determined according to the procedures recommended by EURACHEM/CITAT Guide Quantifying Uncertainty in Analytical Measurement.
In the estimation of the uncertainty it is necessary to take each source and treat it separately to assess their contribution. When this component is expressed as a standard deviation is known as standard uncertainty, and is indicated as u(y).
As can be seen in equation 1, the “y” value of a combined standard uncertainty, UC(y), is calculated based on the uncertainty of the independent parameters p, q, r,…; e.g., u(y=q)
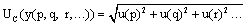 (1)
(1)
Where y(p, q, r…) is a function of the individual parameters that cause uncertainty such as stock and working solution preparation, sample preparation, precision, accuracy, calibration curve, etc.
The expanded uncertainty (Ue) provides an interval where the measured value is expected to lie with an appropriate level of confidence. As shown in equation 2, Ue value is calculated by multiplying the combined uncertainty uc(y) by a coverage factor "k". The value selection for "k" is based on the level of confidence desired, and in this paper the coverage factor chosen was 2, corresponding to a confidence level of 95%.
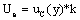 (2)
(2)
During the estimation of the expanded uncertainty for each ECs of the presented method six sources of uncertainty were taken into account: stock solution preparation (u1) and working solution preparation (u2), sample preparation (u3), precision (u4), accuracy (u5) and calibration curve (u6).
In the stage of stock solution preparation, the purity of analytes is a common source of uncertainty. All ECs standards have a purity of 98%, with a tolerance of ± 2%. To determine the individual uncertainties the value of tolerance established by the manufacturer (a) should be treated as a rectangular distribution so that “d=3” in equation 3. In micropipettes and flasks manipulation “d=6”, uncertainty is obtained by dividing the manufacturer´s specification by 6 to convert triangular deviation limits to standard deviation, using equation 3. On the other hand, the weighing of analyte is another element of uncertainty. In this case the manufacturer provides data of u(x)=0.1 mg.
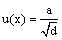 (3)
(3)
With respect to volumetric uncertainty associated with the glassware, used in the preparation of stock and working solution, and in sample treatment (SPE), was calculated considering the coefficient of volume expansion of the liquid being employed at working temperature (equation 4).
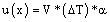 (4)
(4)
Where V=working volume, ΔT=difference between working temperature and calibration temperature and α=coefficient of volume expansion of the liquid employed.
In the estimation of combined uncertainty (uc), the relationship between the individual uncertainties u(y) value "y" and the uncertainty of the individual parameters in each stage is evaluated with equation 5, where (u(x)/x) is the individual uncertainty (volumetric, weighing, analyte purity, etc) expressed as relative standard deviation.
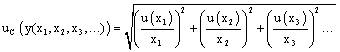 (5)
(5)
In the estimation of the uncertainty associated with the precision and accuracy, the number of repetitions is an important factor. Both estimations are performed for each analyte in the same way. For these estimations the standard deviation of the normalized differences (SDND) for each analyte is divided by 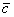 at a determined concentration (Equation 6). In this study the concentration chosen was the middle concentration of the calibration curve (0.2 mg/L).
at a determined concentration (Equation 6). In this study the concentration chosen was the middle concentration of the calibration curve (0.2 mg/L).
 (6)
(6)
With regard to the uncertainty associated with the calibration curve, the curve equation for each analyte is an important factor for its evaluation. The signal average (absorbance measurements) of the replicates was calculated at 0.2 mg/L. Equations 7-9 shows the individual calculation of uncertainty for each analyte.
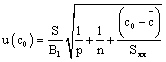 (7)
(7)
Where S represent the residual standard deviation (equation 8), B1 represent the calibration curve slope, co is the evaluated concentration (0.2 mg/L), p is the number of repetitions to determine c0, n is the number of measurements for all the concentrations in calibration curve , is the average concentration of the calibration point, and Sxx was calculated using Equation 9. B0 is the intercept of calibration curve for each analyte, A is the signal of the concentration of the calibration point (j….n) and cj is the concentration of the calibration point (j….n).
 (8)
(8)
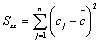 (9)
(9)
SFC-DAD optimization
The chromatographic conditions were optimized to separate each EC with good resolution within a reasonable analysis time. To achieve this, various organic modifiers are usually added to the supercritical fluid CO2, which increases the solubility of polar compounds, improving peak shape and sensitivity of the method. In this method AcN was used as organic modifier added to the mobile phase. Optimization of chromatographic separation was carried out on a Viridis BEH 2-EP column injecting a methanolic solution mixture of ECs at 1 mg/L. Using the conditions described in 2.3 section, the baseline separation was achieved in less than 10 minutes (Figure 1), with resolution (Rs) values higher than 2.3 and symmetry peaks values near 1, with exception of GBD.

Figure 1: SFC-DAD chromatogram of a standard solution at 1 mg/L.
The effect of temperature (35 and 40°C) and back-pressure regulator (BPR) pressure (1500 and 2000 psi) was evaluated using the optimum gradient separation. These parameters may affect chromatographic separation and sensitivity by changing the density and viscosity of the mobile phase. After establishing the optimal conditions of temperature and BPR pressure at 40°C and 2000 psi, respectively, the effect of flow-rate at 1 and 1.4 mL/min were tested. At a flow-rate of 1 mL/ min, a significant decrease resolution of 17EE and BPA was observed. Therefore, the flow-rate was set at 1.4 mL/min. The wavelength for ECs detection was set at 215 nm.
SPE optimization
For optimization of SPE three different SPE cartridges (HF Bond Elut-C18, Bond Elut-C18OH and Discovery DSC-18) were compared. The working protocol was as follows: The cartridge was conditioned with 3 mL of MeOH and then 6 mL of ultra-pure water. Subsequently, a volume of 150 mL of water sample (WS) spiked with a ECs standard mixture of 5 mg/L was placed in a 250 mL flask and passed through the cartridge at 4 mL/min flow-rate. ECs were eluted with 3 mL of MeOH at 1 mL/min flow-rate, and injected into the SFC-DAD system. As can be seen in Table 1, the best results were obtained using Bond Elut- C18OH cartridges and consequently selected for further optimization.
| Compound | Recovery (%) | ||
|---|---|---|---|
| Bond Elut-C18OH | HF Bond Elut-C18 | Discovery DSC-18 | |
| TCS | 87.8 | 87.9 | 12.8 |
| DIU | 74.0 | 62.4 | 37.6 |
| 17EE | 81.2 | 67.6 | 24.2 |
| BPA | 60.4 | 51.2 | 16.0 |
| 17E | 86.0 | 73.4 | 0 |
| CBM | 64.7 | 55.3 | 26.3 |
| GBD | 65.1 | 58.6 | 21.2 |
Table 1: Recoveries using three different SPE cartridges.
The influence of sample flow-rate (4, 6 and 8 mL/min) and pH (4.0, 5.5 and 7.0), washing solution (0, 5 and 15% MeOH, v/v) and elution solvent volume (4, 3, 2, and 1 mL MeOH) on the ECs extraction in WS was evaluated. There were no significant influence when varying sample flow-rate (RSD<1.5%) and sample pH (RSD<3.7%). The effect of washing solution was evaluated by visual inspection and no change in signals was observed. Respect to the elution solvent volume, it was observed that by using of 3 mL MeOH higher responses were obtained comparing with 4, 2 and 1 mL MeOH.
Therefore, the optimal conditions for SPE step were: sample flowrate of 8 mL/min at pH 5.5, no washing step was applied and elution solvent volume was 3 mL MeOH.
Method validation
The method selectivity was evaluated by qualitative comparison of the chromatograms obtained from the standards to 1 mg/L (Figure 1) and WS added with 5 mg/L (Figure 2a) by performing the procedure described in 2.3 and 2.2 section. UV spectrum of each peak detected was compared with the previously stored of the respective standard. As can be seen in Figure 2b, blank sample chromatogram has no signals caused by impurities. Thus, this method is suitable for analysis of ECs in WS.

Figure 2: SFC-DAD chromatogram of a (a) WS spiked at 5 mg/L and (b) blank WS.
The method linearity was performed using a calibration curve of 8 concentrations: 0.014, 0.02, 0.04, 0.1, 0.20, 0.40, 0.60 and 2 mg/L, according to the methodology described in the 2.3 section. The results were evaluated by linear regression where the correlation coefficients (r) for all ECs were higher than 0.9997.
The precision and accuracy of the method were evaluated by triplicate analysis at a concentration of 0.2 mg/L. With regard to precision, evaluated by intra-day repeatability, triplicate determinations were carried out on three consecutive days, with relative standard deviations (RSD) less than 9.2%, as shown in Table 2. Referring to accuracy, expressed as recovery percentages (%R), were found in a range of 94.5 to 103.1% (Table 2), which are within the acceptance criteria.
| Compound | Intra-day Precisiona (% RSD) | Accuracya % Recovery (RSD) | LODb (μg L-1) | LOQb (μg L-1) |
|---|---|---|---|---|
| TCS | 5.5 | 101.0 (6.0) | 0.40 | 1.22 |
| DIU | 5.5 | 94.7 (5.8) | 0.43 | 1.31 |
| 17EE | 7.0 | 103.1 (7.2) | 1.59 | 4.83 |
| BPA | 6.1 | 102.6 (6.2) | 0.23 | 0.69 |
| 17E | 6.7 | 95.6 (7.0) | 0.71 | 2.15 |
| CBZ | 5.5 | 101.9 (5.7) | 0.10 | 0.31 |
| GBD | 6.0 | 96.0 (6.1) | 1.41 | 4.27 |
aIntra-day precision and recovery were assessed at 0.20 mg/L (n=3)
bLimit of detection (LOD) and quantification (LOQ)
Table 2: Validation parameters: Intra-day precision, accuracy, and limit of detection (LOD) and quantification (LOQ) for ECs using WS.
For the evaluation of LOD and LOQ 20 blank WS were analyzed and the standard deviation (SD) of responses in the retention time of each analyte was calculated. These values of SD were divided by the slope of the calibration curve of each analyte and multiplied by 3.3 and 10 to calculate LOD and LOQ, respectively. The values obtained are shown in Table 2.
Measurement uncertainty
Six sources of uncertainty were taken into account: the preparation of the stock solution at 1000 mg/L (u1), the preparation of working solution at 100 mg/L (u2), the sample preparation step using SPE (u3), uncertainty associated with precision (u4), uncertainty associated with accuracy (u5) and uncertainty associated with the linearity regression (u6). Uncertainty of volumetric flasks and automatic pipettes (associated with u1, u2 and u3) were calculated using the tolerance values provided by the manufacturer. Uncertainty associated with precision (u4) and accuracy (u5) were evaluated taking into account the middle point of the calibration curve (0.2 mg/L, n=20). For linearity regression uncertainty (u6) signals, calibration curve data and number of repetitions were used. The combined uncertainty (Uc) of this method was calculated using the Equation 1. The expanded uncertainty (Ue) is obtained by applying a coverage factor of k=2, corresponding to a 95% confidence level around the results where the measurand may lie.
As shown in Table 3, method expanded uncertainty (Ue) were in the 8.30-10.80% range, being the highest to TCS and the lowest to 17E. The individual expanded uncertainties values for preparation of stock (u1) and working (u2) solution for all analytes, u1 and u2 were 1.20 and 0.59 respectively due to their were obtained based on the manufacturer specifications data. In similar way, the uncertainty for all ECs in the stage of sample preparation (u3) was 1.45. During this stage, the use of volumetric glassware had the higher contribution. Uncertainties associated with accuracy (u5) and linearity regression (u6) had the major contribution to Uc and Ue for all compounds.
| Compound | Precision,%(u4) | Accuracy,%(u5) | Linearity regression,%(u6) | Uc (%) | Ue, k=2,(%) |
|---|---|---|---|---|---|
| TCS | 1.88 | -3.13 | 3.46 | 5.40 | 10.80 |
| DIU | 1.66 | -3.14 | 2.92 | 5.00 | 10.00 |
| 17EE | 1.53 | -2.18 | 2.99 | 4.46 | 8.92 |
| BPA | 0.19 | -2.30 | 2.86 | 4.17 | 8.34 |
| 17E | 0.28 | -2.35 | 2.78 | 4.15 | 8.30 |
| CBZ | 2.43 | -2.50 | 2.67 | 4.81 | 9.62 |
| GBD | 2.03 | -2.41 | 2.63 | 4.55 | 9.10 |
Table 3: Uncertainties estimated in each step of ECs analysis for each compound. Uncertainty associated with stock solution preparation at 1000 mg/L (u1), working solution preparation at 100 mg/L (u2) and sample treatment (u3) is 1.20, 0.59 and 1.45 %, respectively.
As can be seen in Table 3, precision uncertainties are in the 1.5- 2.5% range, with exception of BPA (0.19%) and 17E (0.28%). Accuracy uncertainties are around-2.4%, with exception of TCS (3.13%) and DIU (3.14%). Linearity regression uncertainties are in the 2.6-3.0%, with exception of TCS (3.46%). Expanded uncertainty values demonstrate that with a confidence level of 95%, this method is efficient and suitable for the determination of the target ECs in water samples.
Method feasibility
It was performed in 7 different surface WS. For determination, UV spectra of the peaks found in the sample were compared with the corresponding previously determined standards. The results obtained shows that TCS could be quantified at concentrations of 1.3 and 1.2 μg/L in WS4 and WS5, respectively, but in WS1, WS3, WS6, and WS7 were lower than LOQ. BPA concentration in WS4 was 0.70 μg/L, but lower than LOQ in WS2. DIU, 17E, 17EE, CBZ and GBD were not detected in any sample.
sensitive and rapid SPE-SFC-DAD method for the determination of TCS, DIU, 17EE, 17E, CBZ and GBD in WS was developed. Chromatographic separation was performed on a Viridis BEH-2-EP column, using CO2 and AcN as the mobile phase in gradient mode. SPE was carried out using Bond Elut-C18OH cartridges. The method was validated according to international validation guidelines, obtaining recoveries higher than 94%, with LOD and LOQ in a range of 0.10- 1.59 mg/L and 0.31-4.83 mg/L, respectively. For all compounds, the uncertainty associated with accuracy and linearity regression was the main contribution to the Uc and Ue. Expanded uncertainty (Ue) values were lower than 10.80% this implies that the proposed method is reliable for the determination of the target ECs in water samples.
Salvatierra-Stamp thanks Consejo Nacional de Ciencia y Tecnologia (CONACyT) Mexico for the grant provided. This study was funded by CONACyT Mexico grand project 254184.