Organic Chemistry: Current Research
Open Access
ISSN: 2161-0401
ISSN: 2161-0401
Research Article - (2016) Volume 5, Issue 1
New benzimidazole derivatives and their N-substituted acyclic nucleoside analogues were prepared. The synthesized compounds were tested for their antimicrobial activity against Escherichia coli, Staphylococcus aureus, Micrococcus, Salmonella typhi and Salmonella para typhi. The synthesized compounds were tested also against fungi species such as Aspergillus flavus, Aspergillus fumigates, Aspergillus ochraceus and Candida albicans. Most of tested compounds exhibited moderate to high antimicrobial activity while few compounds were found to exhibit little or no activity against the tested microorganisms.
Keywords: Benzimidazole derivatives; Sugar hydrazones; Acyclic nucleosides; Antimicrobial activity
Imidazoles are common scaffolds in highly significant biomolecules, including biotin, the essential amino acid histidine, histamine, the pilocarpine alkaloids [1] and other alkaloids, which have been shown to exhibit interesting biological activities such as antimicrobial, anticryptococcal, inhibition of nitric oxide synthase and cytotoxic activities [2]. Imidazole derivatives have also been found to possess many pharmacological properties and are widely implicated in biochemical processes. Members of this class of diazoles are known to possess NO synthase inhibition [3], antibiotic [4], antifungal [5-8] and neuropeptide Y antagonistic activities [9]. In addition, these heterocycles include several inhibitors of p38 MAP kinases [10-13] which are thought to be involved in a variety of inflammatory and immunological disorders, and some derivatives such as mitronidazole, etomidate and ketoconazole which have found application in drug therapy [14]. Recently certain imidazole based compounds were reported to possess antimicrobial activities [15]. Further recent literature revealed that N1-substitution of imidazoles improves the antibacterial activity [16]. Among the five membered nitrogen heterocycles, the 1,3,4-oxadiazoles are associated with broad spectrum of biological activities [17-19]. Their derivatives have been known to possess antibacterial [20], antimicrobial [21], insecticidal [22], herbicidal, fungicidal [23], anti-inflammatory [24], hypoglycemic [25] characteristics, antiviral [26], and anti-tumour activities [27]. On the other hand, the acyclic C-nucleoside analogues possess a wide range of biological properties, including antibiotic, antiviral, and anti-tumour activities [28-37]. Consequently, newly synthesized compounds containing imidazole and 1,3,4-oxadiazole moieties as well as their substituted sugar derivatives will be expected of enhanced biological activities. Owing to the above facts and our interest in the attachment of carbohydrate residues to newly synthesized heterocycles [38-40] searching for potent leads as antimicrobial agents, our aim is the synthesis and antimicrobial evaluation of new substituted 5-nitroimidazol and their acyclic nucleoside analogues.
Melting points were determined with a Kofler block apparatus and are uncorrected. The IR spectra were recorded on a Perkin-Elmer model 1720 FTIR spectrometer for KBr discs. NMR spectra were recorded on a Varian Gemini 200 NMR Spectrometer at 300 MHz for 1H NMR with TMS as a standard. The progress of the reactions was monitored by TLC using aluminum silica gel plates 60 F 245. Elemental analyses were performed at the Microanalytical data centre at Faculty of science, Cairo University, Egypt.
1-Ethoxycarbonylmethyl-2-methylbenzimidazole (172)
Ethylcholoroacetate (1.4 g, 0.01 mol) was added to a solution of 2-methylbenzimidazol (171) (1.32 g, 0.01 mol) in dry acetone (30 ml) and anh. Potassium carbonate (1.38 g, 0.01 mol). The reaction mixture was heated under reflux for 5 h, poured on crushed-ice, filtered off, and recrystallized from ethanol to give 172 as a white needles (1.82 g, 83%); m.p.=115-117°C. IR spectrum (KBr), ν, cm-1: 1730 (C=O), 1455 (CH2), 1375 (CH3). 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz): 1.32 (t, 3H, J=5.6 Hz, CH3CH2), 1.87(s,3H, CH3), 4.20 (q, 2H, J=5.6 Hz, CH3CH2), 5.08 (s, 2H, NCH2), 7.12- 8.14(m, 4H, Ar-H). Mass spectrum, m/z (I, %): 218[M+] (20).
1-(2-Methylbenzimidazol-1-yl) acetic acid hydrazide (173)
A solution of the respective ester 172 (2.18 g, 0.01 mol) in absolute ethanol (30 ml) and hydrazine hydrate (1.5 g, 0.03 mol) was refluxed for 3 h. The solvent was removed under reduced pressure and the remaining precipitate was collected, dried, and recrystallized from ethanol to afford 173 as a pale yellow powder (1.7 g, 83%); m.p.=88- 90°C. IR spectrum (KBr), ν, cm-1: 1637 (C=O), 3351 (NH), 3567 (NH2). Mass spectrum, m/z (I, %): 204 [M+] (100).
General procedure for the synthesis of Sugar (2-methylbenzimidazol-1-yl)acetylhydrazones 178-181
Hydrazide 173 (0.65 g, 0.032 mol) in ethanol (10 ml) was added to a stirred solution of the respective monosaccharide (0. 032 mol) in water (1 ml) and glacial acetic acid (1 ml). The mixture was heated under reflux, the excess of ethanol was removed under reduced pressure and the residue was triturated with (10 ml) diethyl ether to produce compounds as brown viscous material to afford the corresponding sugar hydrazones 178-181.
L-(-)-Arabinose(2-methylbenzimidazol-1-yl acetylhydrazone (178)
Pale brown gum (0.75 g, 70%); IR spectrum (KBr), ν, cm-1: 3350 (OH), 3469 (NH). 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz):1.00 (s,1H, CH3), 3.25-3.74 (m, 5H, H-2′, H-3′, H-4′, H-5′, H-5′′), 4.45(brs, 2H, 2xOH), 4.60 (s, 2H, CH2), 4.96 (brs, 2H, 2xOH), 7.09(d, 1H, J=2.5 Hz, H-1′), 7.12-7.45 (m, 4H, Ar-H), 9.63 (brs, 1H, NH).
D-(+)-Xylose(2-methylbenzimidazol-1-yl)acetylhydrazone (179)
Pale brown gum (0.82 g, 76%); IR spectrum (KBr), ν, cm-1: 3373 (OH), 3426 (NH). 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz): 0.96 (s,1H, CH3), 3.21-3.70 (m, 5H, H-2′, H-3′, H-4′, H-5′, H-5′′), 4.43(bs, 2H, 2xOH), 4.65 (s, 2H, CH2), 5.22 (bs, 2H, 2xOH), 7.09(d, 1H, J=2.5 Hz, H-1′), 7.12-7.45 (m, 4H, Ar-H), 9.63 (brs, 1H, NH).
D-(+)-Galactose(2-methylbenzimidazol-1-yl acetylhydrazone (180)
Pale brown gum (0.95 g, 81%); IR spectrum (KBr), ν, cm-1: 3295(OH), 3402 (NH). 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz):1.00 (s,3H, CH3), 3.24-3.85 (m, 5H, H-3′, H-4′, H-5′, H-6′, H-6′′), 4.19 (m, 1H, H-2′), 4.42 (bs, 1H, OH), 4.55 (bs, 2H, 2xOH), 4.62 (s, 2H, CH2), 5.00 (bs, 2H, 2xOH), 7.11(d, 1H, J=2.5 Hz, H-1′), 7.38-7.94 (m,4H, Ar-H), 8.88(brs,1H,NH). Mass spectrum, m/z (I, %): 366[M+] (100).
D-(+)-Mannose(2-methylbenzimidazol-1-yl)acetylhydrazone (181)
Pale brown gum (0.85 g, 72%); IR spectrum (KBr), ν, cm-1: 3295(OH), 3402 (NH). 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz):1.00 (s,3H, CH3), 3.20-3.65 (m, 5H, H-3′, H-4′, H-5′, H-6′, H-6′′), 4.19 (m, 1H, H-2′), 4.44 (bs, 1H, OH), 4.55 (bs, 2H, 2xOH), 4.65 (s, 2H, CH2), 5.00 (bs, 2H, 2xOH), 7.11(d, 1H, J=2.5 Hz, H-1′), 7.38- 7.94 (m,4H, Ar-H), 9.00(brs,1H,NH).
General procedure for the synthesis of sugar of tetra-O-acetyl- and penta-O-acetyl(2-methylbenzimidazol-1-yl)acetylhydrazones 183-187
Acetic anhydride (3.06 g, 0.03 mol) was added to a solution of sugar hydrazones 178-181 (0.001 mol) in pyridine (7 ml) with stirring at room temperature for overnight. The mixture was cooled and poured on crushed ice. Hydrochloric acid was added with stirring until the odor of pyridine was removed and extracted by chloroform. The product was separated in pure form as brown viscous material to afford 182-185.
2,3,4,5-Tetra-O-acetyl-L-(-)-arabinose(2-methylbenzimidazol- 1-yl)acetylhydrazone (182)
Pale yellow gum (0.54 g, 60%); 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz): 0.87 (s,3H, CH3), 1.99, 2.01, 2.10, 2.49 (4s, 12H, 4xCH3CO), 3.40,3.41 (2m, 2H, H-5′, H-5′′), 4.23 (m, 1H, H-4′), 4.60 (s, 2H, CH2), 4.97 (m, 1H, H-3′), 5.73 (m, 1H, H-2′), 7.26 (d, 1H, J=2.5 Hz, H-1′), 7.38-7.94 (m,4H, Ar-H), 9.43(brs,1H,NH).
2,3,4,5-Tetra-O-acetyl-D-(+)-xylose(2-methylbenzimidazol-1-yl)acetylhydrazone (183)
Pale yellow gum (0.50 g, 66%), m.p.=>300°C. NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz): 0.87 (s,3H, CH3), 1.99, 2.01, 2.10, 2.49 (4s, 12H, 4xCH3CO), 3.40,3.41 (2m, 2H, H-5′, H-5′′), 4.23 (m, 1H, H-4′), 4.60 (s, 2H, CH2), 4.97 (m, 1H, H-3′), 5.73 (m, 1H, H-2′), 7.26 (d, 1H, J=2.5 Hz, H-1′), 7.38-7.94 (m,4H, Ar-H), 9.43(brs,1H,NH).
2,3,4,5,6-Penta-O-acetyl-D-(+)-galactose(2-methylbenzimidazol- 1-yl)acetylhydrazone (184)
Pale brown gum (0.53 g, 67%); 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz): 0.89(s,3H, CH3), 1.98, 2.02, 2.04, 2.07, 2.48 (5s, 15H, 5xCH3CO), 3.40,3.42 (2m, 2H, H-6′,H-6′′), 4.12 (m, 1H, H-5′), 4.14(s, 2H, CH2), 5.00 (m, 1H, H-4′), 5.30 (m, 1H, H-3′), 5.49 (m, 1H, H-2′), 7.27 (d, 1H, J=2.5 Hz, H-1′),7.68-7.69 (m, 4H, Ar-H), 9.43 (brs, 1H, NH).
2,3,4,5,6-Penta-O-acetyl-D-(+)-mannose(2-methylbenzimidazol- 1-yl)acetylhydrazone (185)
Pale brown gum (0.60 g, 77%); 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz): 0.91(s,3H, CH3), 1.99, 2.00, 2.04, 2.10, 2.48 (5s, 15H, 5xCH3CO), 3.40,3.42 (2m, 2H, H-6′,H-6′′), 4.10 (m, 1H, H-5′), 4.52(s, 2H, CH2), 5.07 (m, 1H, H-4′), 5.30 (m, 1H, H-3′), 5.49 (m, 1H, H-2′), 7.32 (d, 1H, J=2.5 Hz, H-1′),7.55-7.66 (m, 4H, Ar-H), 9.33 (brs, 1H, NH).
General procedure for the synthesis of 4-acetyl-5-(tetra-and penta-O-acetylalditolyl)-2-(2-methylbenzimidazol-1-yl)-1, 3, 4-oxadiazolines 186- 189
A solution of sugar hydrazones 178-181 (0. 01 mol) in acetic anhydride (5 ml) was heated at 100°C for 5 h. The resulting solution was poured onto crushed-ice and the product that separated out was filtered off, washed with a saturated solution of sodium bicarbonate followed by water, and then dried. The products were recrystallized from ethanol to give 186-189.
4-Acetyl-5-(1,2,3,4-tetra-O-acetyl-L-arabinotetritolyl)-2-(2- methylbenzimidazol-1-yl)-1,3,4-oxadiazoline (186)
Pale yellow gum (0.53 g, 65%); IR spectrum (KBr), ν, cm-1: 1741 (COCH3),1650(C=N). 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz): 0.89(s,3H, CH3), 1.83, 1.90, 2.01, 2.10, 2.48 (5s, 15H, 5xCH3CO), 3.85 (s, 2H, CH2), 4.01, 4.09 (2m, 2H, H-4′,H-4′′), 4.97 (m, 1H, H-3′), 5.18 (m, 1H, H-2′), 5.40 (dd, 1H, J=3.2, 6.2 Hz, H-1′),5.89 (d, 1H, J=8.8 Hz, oxadiazoline H-5), 7.68-7.69 (m, 4H, Ar-H).
4-Acetyl-5-(1,2,3,4-tetra-O-acetyl-D-xylotetritolyl)-2-(2-methylbenzimidazol- 1-yl)-1,3,4-oxadiazoline (187)
Pale yellow gum (0.45 g, 55%); IR spectrum (KBr), ν, cm-1: 1738 (COCH3), 1650 (C=N). 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz): 0.89(s,3H, CH3), 1.91, 1.95, 1.97, 1.98, 2.11 (5s, 15H, 5xCH3CO), 3.89 (s, 2H, CH2), 4.00, 4.12 (2m, 2H, H-4′,H-4′′), 5.07 (m, 1H, H-3′), 5.22 (m, 1H, H-2′), 5.47 (dd, 1H, J=3.2, 6.2 Hz, H-1′),5.89 (d, 1H, J=8.8 Hz, oxadiazoline H-5), 7.68-7.69 (m, 4H, Ar-H).
4-Acetyl-5-(1,2,3,4,5-penta-O-acetyl-D-galactopentitolyl)-2- (2-methylbenzimidazol-1-yl)-1,3,4-oxadiazoline (188)
Pale yellow gum (0.62 g, 73%); IR spectrum (KBr), ν, cm-1: 1738 (COCH3), 1674 (C=N). 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz): 0.89(s,3H, CH3), 1.90, 1.91, 1.95, 1.97, 1.98, 2.11 (6s,18H,6xCH3CO), 3.89 (s, 2H, CH2), 4.05, 4.12 (2m,2H,H-5′,H-5′′), 4.98 (m,1H,H-4′), 5.23 (m, 1H, H-3′), 5.33 (m,1H,H-2′), 5.43 (dd,1H, J=3.2, 6.2 Hz,H-1′),5.89 (d, 1H, J=8.8 Hz,oxadiazoline H-6), 7.68-7.69 (m, 4H, Ar-H).
4-Acetyl-5-(1,2,3,4,5-penta-O-acetyl-D-mannopentitolyl)-2- (2-methylbenzimidazol-1-yl)-1,3,4-oxadiazoline (189)
Pale yellow gum (0.58 g, 69%); IR spectrum (KBr), ν, cm-1: 1742(COCH3), 1679 (C=N). 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz): 0.89(s,3H, CH3), 1.90, 1.91, 1.95, 2.03, 2.10, 2.13 (6s, 18H, 6xCH3CO), 3.95 (s, 2H, CH2), 4. 0.89(s,3H, CH3), 11, 4.17 (2m, 2H, H-5′,H-5′′), 4.92 (m, 1H, H-4′), 5.19 (m, 1H, H-3′), 5.31 (m, 1H, H-2′), 5.43 (dd, 1H, J=3.2, 6.2 Hz, H-1′),5.89 (d, 1H, J=8.8 Hz, oxadiazoline H-6), 7.68-7.69 (m, 4H, Ar-H).
2-[(2-methylbenzimidazol-1-yl)methyl]-1,3,4-oxadiazole-2- thiol (194)
To a solution of (173) (4.8 g, 0.02 mole) in ethanol (50 mL) was added a solution of potassium hydroxide (1.12 g, 0.02 mole) in water (2 mL) and carbon disulphide (5 mL). The solution was heated under reflux for 15 h. The solvent was evaporated and the residue was dissolved in water, filtered, and acidified with dilute hydrochloric acid. The precipitate was filtered off, washed with water and recrystallized from ethanol. Yield 80%; mp=200-202°C; IR (KBr): ν 3415 (NH), 1622 cm-1 (C=N). 1H NMR (DMSO-d6, 300 MHz): δ1.00 (s,1H, CH3), 4.58 (s, 2H, CH2), 7.97-8.22 (m, 4H, Ar-H), 12.80 (s, 1H, SH). m/z=246 [M+].
2-(Methylthio)-2-[(2-methylbenzimidazol-1-yl)methyl]- 1,3,4-oxadiazole (195)
To a solution of (194) (2.46 g, 0.01 mole) and potassium hydroxide (0.56 g, 0.01 mole) in a mixture of water (25 mL) and ethanole (10 mL), was added the methyl iodide (0.01 mole). The solution was stirred at room temperature for 4 h. The resulting precipitate was filtered off and crystallized from ethanol to give 195. Yield 71%; mp=87-89°C; IR (KBr): ν 1622 cm-1 (C=N). 1H NMR (DMSO-d6, 300 MHz): δ 0.9(s, 3H, CH3), 2.65 (s, 1H, SCH3), 5.55 (s, 2H, CH2), 7.98-8.22(m, 4H, Ar-H).
2-Hydrazinyl-2-[(2-methylbenzimidazol-1-yl)methyl]-1,3,4- oxadiazole (196)
A mixture of (195) (0.01 mole) EtOH (8 mL), and N2H4.H2O (1 g, 0.02 mole) was refluxed for 4 h and the solvent was removed under reduced pressure. The remaining precipitate was collected, dried, and recrystallized from EtOH to afford compound 196. Yield 74%; m.p.=174-176°C; IR (KBr): ν 1620 cm-1 (C=N). 1H NMR (DMSO-d6, 300 MHz): δ1.00(s,3H, CH3), 5.58 (s, 2H, CH2), 5.80 (s, 2H, NH2), 7.95- 8.22(m, 4H, Ar-H),9.45(brs, 1H, NH).
General procedures for the reaction of (196) with aromatic aldehydes to afford Schiff′s bases 200-202
A solution of 196 (2.44 g, 0.01 mol), an aromatic aldehyde (0.01 mol) in abs. ethanol (30 ml) and glacial acetic acid (1 ml) was refluxed for 4-6 h (TLC). The solvent was evaporated under reduced pressure and the residue was filtered off and recrystallized from ethanol to afford 200-202.
N′-(Benzylidene)-2-[(2-methylimbenzimidazol-1-yl)methyl] -1,3,4-oxadiazole (200).
yellow powder (2.78 g, 83%); m.p.=270-272°C. 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz):0.94(s,3H, CH3), 4.51 (s, 2H, CH2), 7.80 (s, 1H, CH), 7.86-8.01 (m, 8H, Ar-H), 9.93 (brs, 1H, NH).
N′-(4-Chlorobenzylidene)-2-[(2-methylimbenzimidazol- 1-yl)methyl]-1,3,4-oxadiazole (201)
White powder (3.24 g, 88%); m.p.=145-147°C. 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz):0.94(s, 3H, CH3), 4.55 (s, 2H, CH2), 8.00 (s, 1H, CH), 8.42-8.64 (m, 8H, Ar-H), 9.93 (brs, 1H, NH). Mass spectrum, m/z (I, %): 366 [M+] (60).
N′-[5-(Methylfuran-2-yl)methylene]-2-[(2-methylimbenzimidazol- 1-yl)methyl]-1,3,4-oxadiazole (201).
White powder (3.10 g, 92%); m.p.=299-300°C. 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz):0.95(s,3H, CH3), 2.32 (s, 3H, CH3), 4.65 (s, 2H, CH2), 6.24 (s, 1H, H-4-furan), 6.79 (s, 1H, H-3- furan), 7.22-7.55 (m, 4H, Ar-H), 8.22 (s, 2H, CH=N,),9.96 (brs, 1H, NH). Mass spectrum, m/z (I, %): 336 [M+] (23).
General procedure for the synthesis of sugar hydrazinyl derivatives 205,206
To a well stirred solution of the respective monosaccharides (xylose and galactose) (0.01 mole) in water (2 mL), and glacial acetic acid (0.2 mL) was added to 196 (2.44 g, 0.01 mole) in ethanol (10 mL). The mixture was heated under reflux for 3 h, the resulting solution was concentrated and left to cool. The precipitate formed was filtered off, washed with water and ethanol, then dried and recrystallized from ethanol.
2-D-Xylotetritolylidenehydrazinyl)-2-((2-methylbenzimidazol- 1-yl)methyl)-1,3,4-oxadiazole (205)
Yield 80%; mp=194-196°C; IR (KBr): ν 3445 (OH), 3346 (NH) 1612 cm-1 (C=N). 1H NMR (DMSO-d6, 300 MHz): δ1.05(s,3H, CH3), 3.69 (m, 2H, H-5′, H-5′′), 4.18 (m, 1H, H-4′), 4.37 (dd, 1H, J=2.8 Hz, J=5.8 Hz, H-3′), 4.44 (t, 1H, J=5.8 Hz, H-2′),4.61 (m, 1H, OH), 4.75 (d, 1H, J=6.3 Hz, OH), 4.90 (m, 1H, OH), 4.98 (t, 1H, J=4.5 Hz, OH), 4.97 (s, 2H, CH2), 7.45-7.86(m,4H,Ar-H) 8.10 (d, 1H, H-1′), 11.18 (s, 1H, NH).
2-[2-D-Galactopentitolylidenehydrazinyl)-2-((2-methylbenzimidazol- 1-yl)methyl)-1,3,4-oxadiazole (206)
Yield 82%; mp=182-184°C; IR (KBr): ν 3448 (OH), 3382 (NH), 1612 cm-1 (C=N). 1H NMR (DMSO-d6, 300 MHz): δ 1.05(s,3H, CH3), 3.48 (m, 2H, H-6′, H-6′′), 3.52 (m, 1H, H-5′), 4.18 (m, 1H, H-4′), 4.36 (dd, 1H, J=2.8 Hz, J=5.8 Hz, H-3′), 4.42 (t, 1H, J=5.8 Hz, H-2′), 4.58 (s, 2H, CH2), 4.61 (m, 1H, OH), 4.74 (d, 1H, J=6.3 Hz, OH), 4.91 (m, 1H, OH), 4.97 (t, 1H, J=4.5 Hz, OH), 5.37 (t, 1H, J=4.5 Hz, OH), 7.45-7.86(m,4H,Ar-H), 8.44 (d, 1H, H-1′), 11.14 (s, 1H, NH).
General procedure for the synthesis of per-O-acetyl-sugar hydrazinyl derivatives 209,210
To a solution of compounds 205,206 (0.001 mole) in pyridine (7 mL) was added to acetic anhydride (1.02 g, 0.01 mol). The mixture was stirred at room temperature for 5 h. The resulting solution was poured onto crushed ice, and the product that separated out was filtered off, washed with a solution of sodium hydrogen carbonate followed by water and then dried. The products were recrystallized from ethanol.
2-[2-(2,3,4,5-Tetra-O-acetyl-D-xylotetritolylidene)hydrazinyl]- 2-[(2- methylbenzimidazol-1-yl)methyl]-1,3,4-oxadiazole (209)
Yield 72%; mp=174-176°C; IR (KBr): ν 1740 (C=O), 3282 (NH), 1614 cm-1 (C=N).
2-[2-(2,3,4,5,6-Penta-O-acetyl-D-galactopentitolylidene) hydrazinyl]-2-[(2-methylbenzimidazol-1-yl)methyl]-1,3,4- oxadiazole (210)
Yield 78%; mp=188-190°C; IR (KBr): ν 1740 (C=O), 3282 (NH), 1614 cm-1 (C=N). 1H NMR (DMSO-d6, 300 MHz): δ1.02(s,1H, CH3), 1.93, 1.95, 2.03, 2.07, 2.10 (5s, 15H, 5xCH3CO), 3.98 (dd, 1H, J=11.2 Hz, J=2.8 Hz, H-6′), 4.07 (dd, 1H, J=11.2 Hz, J=3.2 Hz, H-6′′), 4.18 (m, 1H, H-5′), 4.22 (t, 1H, J=7.5 Hz, H-4′), 4.82 (s, 2H, CH2), 5.18 (dd, 1H, J=2.8 Hz, J=6.5 Hz, H-3′), 5.24 (dd, 1H, J=7.5.2 Hz, J=9.5 Hz, H-2′), 7.20 (d,1H, J=7.8 Hz, H-1′), 7.45-7.86(m,4H,Ar-H), 11.20 (s, 1H, NH).
Antimicrobial testing
Antimicrobial screening: The agar diffusion method reported by Cruickshank et al. [41] was used for the screening process. The bacteria and fungi were maintained on nutrient agar and Czapek’s-Dox agar media, respectively. The assay medium flasks containing 50 ml of nutrient agar for bacteria and Czapek’s-Dox agar medium for fungi respectively were allowed to reach 40-50°C to be inoculated with 0.5 ml of the test organism cell suspension. The flasks were mixed well and poured each into a Petri dish (15 × 2 cm) and allowed to solidify. After solidification, holes (0.6 cm diameter) were made in the agar plate by the aid of a sterile cork poorer (diameter 6 mm). The synthesized target compounds were dissolved each in 2 ml DMSO. In these holes, 100 μl of each compound was placed using an automatic micropipette. The Petri dishes were left at 5°C for 1 h to allow diffusion of the samples through the agar medium and retard the growth of the test organism. Plates were incubated at 30°C for 24 h for bacteria and 72 h of incubation at 28°C for fungi. DMSO showed no inhibition zones. The diameters of zone of inhibition were measured and compared with that of the standard and the solvent alone was used as negative control, the values were tabulated. Ciprofloxacin [42,43] (50 μg/ml) and Nystatin [44] (50 μg/ml) were used as standard for antibacterial and antifungal activity respectively. The observed zones of inhibition are presented in Tables 1 and 2.
| Compound | Gram-positive | Gram-positive | Gram-negative | Gram-negative | Gram-negative |
|---|---|---|---|---|---|
| Staphylococcus aureus | Micrococcus | Salmonella typhi | Salmonella para typhi | Escherichia coli | |
| 171 | 0 | 0 | 0 | 0 | 0 |
| 172 | 0 | 0 | 0 | 0 | 220 |
| 178 | 0 | 250 | 130 | 125 | 0 |
| 179 | 150 | 225 | 0 | 0 | 0 |
| 180 | 0 | 0 | 0 | 0 | 0 |
| 181 | 150 | 0 | 125 | 125 | 0 |
| 182 | 80 | 0 | 100 | 0 | 0 |
| 183 | 0 | 130 | weak | 100 | 80 |
| 184 | 125 | 500 | 0 | 225 | 75 |
| 185 | 0 | 0 | 0 | 0 | 0 |
| 186 | 0 | 100 | 130 | 125 | 75 |
| 187 | 0 | 125 | 0 | 0 | 0 |
| 188 | 0 | 240 | 140 | 0 | 100 |
| 189 | 75 | 350 | 125 | 225 | 100 |
| 190 | 75 | 0 | 100 | 110 | 100 |
| 191 | 0 | 110 | 100 | 125 | 75 |
| 192 | 0 | 0 | 0 | 0 | 0 |
| 205 | 110 | 200 | 0 | 0 | 75 |
| 206 | 0 | 0 | 0 | 0 | 0 |
| 207 | 0 | 0 | 0 | 0 | 0 |
| 208 | 0 | 0 | 0 | 0 | 0 |
| 209 | 0 | 0 | 0 | 0 | 0 |
| 210 | 0 | 0 | 0 | 0 | 0 |
| 211 | 0 | 0 | 0 | 0 | 0 |
| 212 | 75 | 350 | 0 | 0 | 125 |
| 213 | 0 | 0 | 0 | 0 | 0 |
| 214 | 75 | 250 | 0 | 0 | 125 |
| 215 | 0 | 0 | 0 | 0 | 0 |
| 216 | 0 | 0 | 0 | 0 | 0 |
| 217 | 0 | 0 | 0 | 0 | 0 |
| 218 | 125 | 225 | 0 | 225 | 130 |
| 219 | 0 | 0 | 0 | o | 0 |
| 220 | 0 | 100 | 0 | 140 | 0 |
| 221 | 0 | 100 | 0 | 0 | 75 |
| 222 | 0 | 0 | 0 | 0 | 0 |
| 223 | 0 | 125 | 0 | 0 | 75 |
| Ciprofloxacin | 125 | 110 | 220 | 225 | 225 |
| DMSO | 0 | 0 | 0 | o | 0 |
Table 1: Minimum inhibitory concentrations (MIC in μg/ml) of the title Compounds against bacteria species. The negative control DMSO showed no activity.
| Compound | Fungi | Fungi | Fungi | Fungi |
|---|---|---|---|---|
| Aspergillus flavus | Aspergillus fumigates | Aspergillus ochraceus | Candida albicans | |
| 171 | 0 | 0 | 0 | 0 |
| 172 | 0 | 0 | 0 | 0 |
| 179 | 0 | 0 | 0 | 0 |
| 181 | 0 | 0 | 0 | 0 |
| 184 | 0 | 0 | 0 | 0 |
| 187 | 0 | 0 | 0 | 0 |
| 189 | 0 | 0 | 0 | 0 |
| 190 | 0 | 0 | 0 | 0 |
| 191 | 0 | 0 | 0 | 0 |
| 205 | 0 | 0 | 0 | 0 |
| 209 | 0 | 0 | 0 | 0 |
| 210 | 0 | 0 | 0 | 0 |
| 211 | 0 | 0 | 0 | 0 |
| 212 | 120 | 500 | 0 | 500 |
| 214 | 0 | 0 | 0 | 0 |
| 216 | 0 | 0 | 0 | 0 |
| 218 | 0 | 0 | 0 | 0 |
| 221 | 0 | 0 | 0 | 110 |
| 222 | 0 | 0 | 0 | 0 |
| 223 | 0 | 0 | 0 | 0 |
| Nystatin DMSO | 250 | 150 | 45 | 225 |
| 0 | 0 | 0 | 0 |
Table 2: Minimum inhibitory concentrations (MIC in μg/ml) of the title Compounds against fungi species. The negative control DMSO showed no activity.
The synthesized compounds were screened in vitro for their antimicrobial activities against Staphylococcus aureus (G+ve bacteria), Micrococcus (G+ve bacteria), Salmonella typhi (G-ve bacteria), Salmonella para typhi (G-ve bacteria) and Escherichia coli (G-ve bacteria) in addition to strains of fungi species Aspergillus flavus, Aspergillus fumigates, Aspergillus ochraceus and Candida albicans which were isolated from milk and milk products. The diameters of zone of inhibition were measured and compared with that of the standard and the solvent alone was used as negative control. The values of minimal inhibitory concentrations (MICs) of the tested compounds are presented in Tables 1 and 2. The MIC values of the results obtained revealed that compounds showed varying degrees of inhibition against the tested microorganisms. The results indicated generally that tested compounds did not show high activity against fungi under test except 194 that exhibited high antifungal activities against (Aspergillus fumigates and Candida albicans) and some antifungal activity against Aspergillus flavus, in addition to 196 that exhibited some antifungal activities against Candida albicans. Also the results indicated that among the compounds tested in antibacterial screening Compound 209 revealed high activities against Micrococcus while Compounds 182, 183, 184, 185, 186 and 187 exhibited good antibacterial activities in addition to Compounds 188, 189, 201, 191 that exhibited appreciable activity against Micrococcus. Compounds 172, 181, 182, 183, 184, 185, 186, 187 and 188 exhibited mild to moderate antibacterial activity against Salmonella para typhi. Compounds 189, 194, 196, 200, 201, 202,205 and 206 exhibited notable antibacterial activity against Salmonella typhi. Compounds 172, 184, 188, 189, 194, 209 and 210 exhibited notable antibacterial activity against Escherichia coli followed by compounds 183, 186, 195 and 206. Compounds 179, 181, 184 and 205 exhibited notable antibacterial activity against Staphylococcus aureus followed by compounds 182, 189, 201, 202 and 205.
2-Metylbenzimidazole (171) was allowed to react with ethylcholoroacetate in dry acetone and in the presence of anhydrous potassium carbonate to afford 1-ethoxycarbonylmethyl-2- methylbenzimidazole (172) in 83% yield. Treatment of 172 with hydrazine hydrate in absolute ethanol at reflux temperature gave the corresponding hydrazide 173 in 83% yield following the reported procedure 146. The structure of 172 was confirmed by IR spectra which showed the presence of characteristic absorption bands corresponding to (CH2) groups at ν 1455 cm-1 and (CO) group at ν 1730 cm-1. 1H NMR showed a signal corresponding to CH3CH2 group as a triplet at δ 1.32 ppm, signal corresponding to CH3CH2 group as a quartet at δ 4.20 ppm, signal corresponding to NCH2 group as a singlet at δ 5.08 ppm, and signal for aromatic at δ 7.12 - 8.14 ppm. Mass spectrum, m/z (I, %): 218[M+] (20). The structure of 173 was confirmed by IR spectra which showed the presence of characteristic absorption bands corresponding to (CO) group at ν 1637 cm-1, (NH) group at ν 3351 cm-1 and (NH2) group at ν 3567 cm-1 in addition confirmed by mass spectra revealed the presence characteristic signals corresponding to the molecular ion peaks corresponding to their molecular formulas. 1-(2-Methylbenzimidazol-1-yl) acetic acid hydrazide (Scheme 1). 173 was allowed to react with L-(-)-arabinose, D-(+)-xylose, D-(+)- galactose and D-(+)-mannose in an aq. ethanolic solution with a catalytic amount of acetic acid, to afford the corresponding sugar N-acylhydrazones 178-181 in 70-81% yields. The structures of these compounds were confirmed by analytical and spectral data. The IR spectra of 178-181 showed the presence of characteristic absorption bands corresponding to (OH) group in the region ν 3295–3350 cm-1. The 1H NMR spectra showed signals of the sugar chain protons at δ 3.24-3.85 ppm, the C-1 methine proton as a doublet in the range δ 7.09- 7.11 ppm, in addition to the aromatic protons in the region δ 7.12-7.94 ppm. Treatment of the sugar hydrazones 178-181 with acetic anhydride in pyridine at room temperature gave the corresponding per-O-acetyl derivatives 182 -185 in 60-77% yields. The 1H NMR spectra of 182-185 showed the signals of the O-acetyl-methyl protons at δ 1.98-2.48 ppm. The rest of the sugar protons appeared in the range δ 3.40-5.73 ppm. The reaction of sugar arylhydrazones with boiling acetic anhydride is well known to give either the corresponding per-O,N-acetyl derivatives or the respective per-O,N-acetyl-1,3,4-oxadiazolin derivatives 147- 149. However, reaction of the sugar hydrazones 178-181 with acetic anhydride at 100°C gave the sugar-substituted 1,3,4-oxadiazoline derivatives 186-189 in 74-81% yields. The IR spectra of 188-192 showed the presence of characteristic absorption bands corresponding to (CO) group in the region ν 1738-1742 cm-1 and bands corresponding to (C=N) group in the region ν 1650-1679 cm-1. The 1H NMR spectra of 186-189 showed signals of the O- and N-acetyl-methyl protons as singlets in the range δ 1.83-2.48 ppm. The rest of the sugar chain protons appeared in the range δ 4.05-5.34 ppm and the aromatic protons as multiplets in the region δ 7.68-7.69 ppm (Scheme 2). When the hydrazide 173 was reacted with carbon disulphide in ethanol in the presence of potassium hydroxide at reflux temperature, it afforded 2-[(2-methylbenzimidazol-1-yl)methyl]-1,3,4-oxadiazole-2-thiol (194) in 80% yield. The 1H NMR spectrum of the 1,3,4-oxadiazole-2- thione showed a signal corresponding to the CH2 group as a singlet at δ 4.58 ppm, Ar-H signals as multiplet at δ 7.97 - 8.48 ppm in addition to the SH signal at δ 12.80 ppm. Alkylation of the 1,3,4-oxadiazolethione 194 with methyl iodide in alkaline medium afforded the corresponding S-methyl derivative 195 in 71% yield. Hydrazinolysis of 195 gave 2-hydrazinyl-2-[(2-methylbenzimidazol-1-yl)methyl]-1,3,4- oxadiazole (196) in 74% yield. The 1H NMR spectra of 195 showed the signals of the methyl group as a singlet which disappeared in the 1H NMR spectrum of 196 in which the NH2 signal appeared at δ 5.80 ppm (Scheme 3). 2-Hydrazinyl-2-[(2-methylbenzimidazol-1-yl)methyl]- 1,3,4-oxadiazole (196) was refluxed with various aromatic aldehydes in ethanol and in the presence of a catalytic amount of acetic acid to afford the corresponding Schiff′s bases 200-202 in 89-95 yields. The structures of 201 and 202 were confirmed by mass spectra which revealed the presence of the characteristic signals corresponding to the molecular ion peaks corresponding to their molecular formulas. The structures of 200, 201, and 202 were confirmed by 1H NMR spectra which showed a signal corresponding to the CH2 group as a singlet at δ 4.51-4.65 ppm, signals of the aromatic protons at δ 7.22-8.64 ppm, a signal corresponding to the CH group (H-4-furan and H-3-furan) as a singlet at δ 6.24 and 6.79 ppm respectively, NH signal at δ 9.93-9.96 ppm, in addition to the CH=N signals as tow singlet at δ 7.80,8.8.00 and 8.22 ppm (Scheme 4). 196 was allowed to react with D-(+)-xylose and D-(+)- galactose in an aq. ethanolic solution with a catalytic amount of acetic acid. The structures of these compounds were confirmed by spectral data. The IR spectra of 205, 206 showed the presence of characteristic absorption bands corresponding to (OH) group in the region ν 3445–3448 cm-1.

Scheme 1: 1-(2-Methylbenzimidazol-1-yl) acetic acid hydrazide.

Scheme 2: 2-[(2-methylbenzimidazol-1-yl)methyl]-1,3,4-oxadiazole-2-thiol (194).

Scheme 3: 2-hydrazinyl-2-[(2-methylbenzimidazol-1-yl)methyl]-1,3,4-oxadiazole (196).

Scheme 4: General procedures for the reaction of (196) with aromatic aldehydes to afford Schiff's bases 200-202.
The 1H NMR spectra showed signals of the sugar chain protons at δ 3.48-4.44 ppm, the C-1 methine proton as a doublet in the range δ 8.10-8.44 ppm, in addition to the aromatic protons in the region δ 7.45-7.86 ppm. Treatment of the sugar hydrazones 205, 206 with acetic anhydride in pyridine at room temperature gave the corresponding per-O-acetyl derivatives 209, 210 in 72, 78% yields. The 1H NMR spectra of 209, 210 showed the signals of the O-acetyl-methyl protons at δ 1.93-2.10 ppm. The rest of the sugar protons appeared in the range δ 3.98-5.24 ppm (Scheme 5).
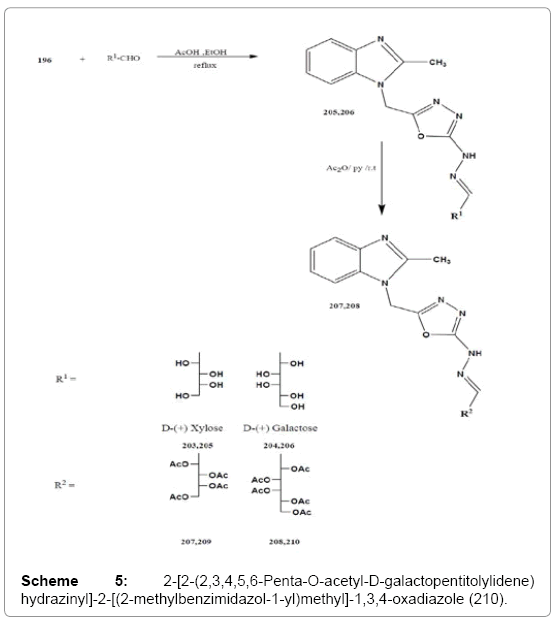
Scheme 5: 2-[2-(2,3,4,5,6-Penta-O-acetyl-D-galactopentitolylidene) hydrazinyl]-2-[(2-methylbenzimidazol-1-yl)methyl]-1,3,4-oxadiazole (210).