Biochemistry & Pharmacology: Open Access
Open Access
ISSN: 2167-0501
ISSN: 2167-0501
Research Article - (2013) Volume 2, Issue 1
Cell proliferation can be regulated by small, aliphatic polyamines, and it is suggested that tumor tissues have significantly higher polyamine levels than surrounding tissues. The major biologically active polyamines present in mammalian cells are putrescine, spermidine, and spermine. The Ornithine Decarboxylase (ODC) catalyzes the decarboxylation of ornithine to produce putrescine which is precursor of polyamine synthesis. We report here the synthesis of 2-Amino-5-(Hydroxyimino) Pentanoic Acid (AHPA), based on the substrate of ODC, L-ornithine, derivatized with oxime functionality. In molecular docking studies, the E-isomer AHPA binds to ODC more favorably than does the
Z-isomer. In addition, the growth of MCF-7 (Michigan Cancer Foundation–7) breast cancer cells in the presence of AHPA was significantly reduced. These results implicate that AHPA a potentialt can be explored as a potential of cancer chemotherapy.
Keywords: Polyamine; Ornithine decarboxylase (ODC); Inhibitors of ODC; MCF-7 (Michigan Cancer Foundation–7)
Cancer is the second most common cause of death for Americans and accounts for nearly 1 of every 4 deaths in the US [1,2]. An estimated 229,060 new cases of invasive breast cancer are expected to occur among women in the US during 2012; about 2,190 new cases are expected in men [1,2]. Cancer is a disease related to uncontrolled growth and spread of abnormal cells [2]. Targeting the polyamine pathway has been studied in possible therapeutic approaches [3]. One of the most promising areas for the development of novel anti-cancer therapeutics is polyamine biosynthesis [4,5]. The finding that inhibitors of polyamine biosynthesis can prevent, or at least limit cancer cell growth [6-9], together with the fact that polyamine concentrations are elevated in multiple cancer tissues [10-12], has made polyamine metabolism a promising target for cancer chemoprevention and therapy. The major biologically active polyamines present in mammalian cells are putrescine, spermidine, and spermine [3]. These molecules are synthesized in sequence starting from ornithine, which is derived from the amino acid arginine through the action of the enzyme arginase (Scheme 1). The first critical step is the synthesis of putrescine via the decarboxylation of ornithine, which is catalyzed by the enzyme Ornithine Decarboxylase (ODC) [13,14]. Subsequent steps involve the production of spermidine through the addition of Decarboxylated S-Adenyosylmethionine (DAM) to the putrescine by spermidine synthase. A second DAM is then added to spermidine to produce spermine [15-17] (Scheme 1).
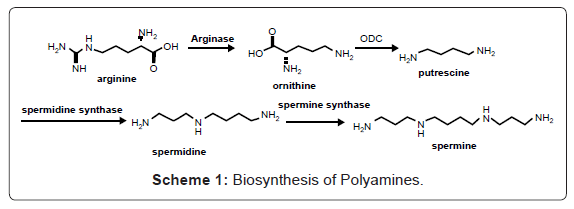
Scheme 1: Biosynthesis of Polyamines.
Difluoromethylornithine (DFMO), an inhibitor of the first enzyme in the mammalian polyamine biosynthetic pathway, ornithine decarboxylase, is approved for use in trypanosomiasis and has shown promise in the therapy of brain tumors [18]. DFMO was originally evaluated as an antitumor agent in the early 1980s, with limited success. Phase I studies suggested a dose of 2.25 g/m2 every 6 h for patients with advanced solid tumors or lymphomas.[19] Phase II studies were carried out with melanoma patients including small cell lung carcinoma, colon cancer, and prostate cancer [20-22]. The drug was generally well tolerated, although significant but infrequent adverse effects including thrombocytopenia (a relative decrease of platelets in blood), transient hearing loss, and osmotic diarrhea were noted. The results of these studies deterred continued evaluation of the drug as an antitumor agent. Although the ODC inhibitor may have significant effects on their respective target enzymes, only one inhibitor, R-Difluoromethylornithine (DFMO), has reached the market. DFMO was originally designed as an antitumor agent, but the drug was not effective enough to further study phase II trials. However, it has been shown to be an effective cure for infection caused by Trypanosoma brucei gambiense, which causes West African sleeping sickness [23,24].
Due to the limitation of use of DFMO, it is necessary to design new possible compounds that disturb the polyamine biosynthetic pathway. The oxime moiety is of great value in investigating binding affinities in arginine biosynthesis due to the geometrical isomers (E/Z) of the oxime functionality and biological activities [25]. The oxime moiety (-C=N-OH) is easily coordinated with metal ions or hydrogen bonds with conserved residues in active sites of various enzymes.
In this present study, we report the design and synthesis of 2-amino- 5-(hydroxyimino) pentanoic acid (AHPA), 6, which contains an oxime functional group. The molecular docking study was conducted to extend Structure-Activity Relationship (SAR) studies based on AHPA with ODC (PDB code 2ON3). Although we synthesize the mixture of E and Z isomers of 2-Amino-5-(Hydroxyimino) Pentanoic Acid (AHPA), we can anticipate that the E isomer has a better binding affinity than the Z-isomer based on a molecular docking study. In addition, the biological evaluation of AHPA with MCF-7 (Michigan Cancer Foundation–7) breast cancer cells was shown more potent when compared with DFMO in vitro. Our initial studies investigated the impact of AHPA on proliferation of human breast cancer cells, and the results indicate that inhibition of ODC greatly impairs the ability of these cells to replicate. Therefore, AHPA may have a considerable potential as a cancer chemopreventive and therapeutic agent.
Chemistry
We designed and synthesized a 2-amino-5-(hydroxyimino) pentanoic acid 6 (AHPA) inhibitor based on the modified substrate of ornithine decarboxylase with oxime functionality. The Compound 6 (APHA) was synthesized using the procedure described in scheme 2.
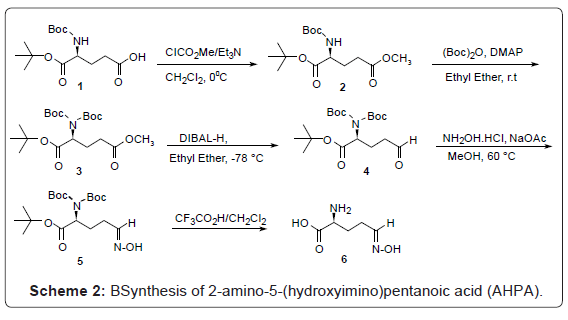
Scheme 2: BSynthesis of 2-amino-5-(hydroxyimino)pentanoic acid (AHPA).
2-Amino-5-(hydroxyimino)pentanoic acid (AHPA), 6, was synthesized using L-glutamic acid derivative (S)-5-(tert-butoxy)-4-[(tert-butoxycarbonyl)amino]-5-oxopentanoic acid 1, as the starting material. Esterification of the compound 1 using methyl chloroformate by treatment with triethylamine and a catalytic amount of 4-dimethylaminopyridine (DMAP) in dry methylene chloride provided the ester derivative 2. Synthesis of di-tert-butyl dicarbonate (di-BOC) 3 was carried out by treatment with di-tert-butyl dicarbonate in the presence of DMAP in methylene chloride. The subsequent reduction of ester derivative 3 was performed with diisobutylaluminium hydride (DIBAL-H) in ether to provide the aldehyde 4 [26]. Oxime derivative 5 was prepared for hydroxylamine hydrochloride in methanol under reflux condition. Complete deprotection of the compound, 5 with trifluoroacetic acid (TFA) in methlyene chloride yielded AHPA 6.
We have investigated the stabilities of geometric isomers in molecular modeling-docking studies to computationally evaluate the fit between the human and Leishmania donovani ODC (PDB code 2ON3) and the E- and Z-isomers of AHPA [27,28]. Calculated binding data are recorded in table 1. When E-AHPA is bound in an extended conformation, the oxime moiety can make hydrogen bonds with conserved enzyme residues, Lys 69, Arg154, and Glu274. When Z-AHPA is bound, the oxime moiety can make a hydrogen bond with only one conserved residue, Asp364. Thus, the greater hydrogen bonding potential of the E-isomer of the oxime moiety suggests a stability preference for this isomer. Force-field based methods can predict the binding free energy of a protein-ligand complex by adding up individual contributions from different types of interactions. Programs for energetic analysis of receptor-ligand interaction based on force-field scoring functions and terms including van der Waals, electrostatics and hydrogen bonds can be available.
| Compound AHPA, 6 | Ntor | ΔGAD4 (kcal/mol) | RMSD (Å) | H-bonding interaction | H-bond distance (Å) |
| E-isomer (trans-) | 3 | -7.92 | 2.347 | HO…HN(Lys 69) HO…HN(Arg154) NH…OH(Glu274) |
2.7 1.9 1.8 |
| Z-isomer (cis-) | 6 | -5.51 | 0.485 | OH…OC(Asp364) | 1.9 |
Table 1: Comparison of the complex of E/Z isomers of 2-Amino-5-(Hydroxyimino) Pentanoic Acid (AHPA) with ODC (PDB code 2ON3) in molecular docking studies
Interestingly, Autodock molecular modeling of the structure of human and Leishmania donovani ornithine decarboxylases [14] (PDB code 2ON3) with geometric isomers (E/Z) of oxime ligands has shown that a high degree of affinity with E isomer rather than Z isomer (Table 1, Figure 1 and Figure 2).
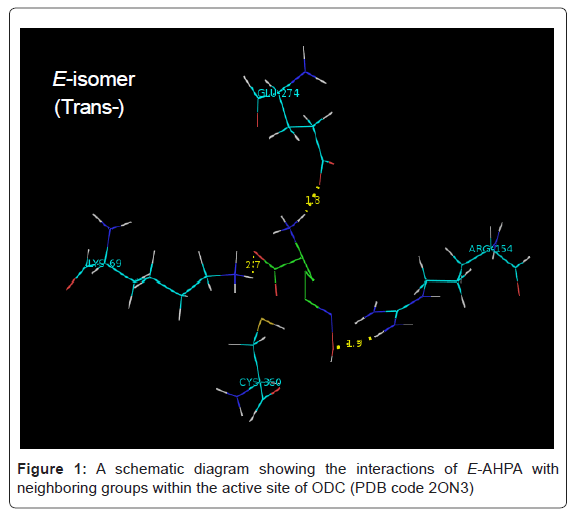
Figure 1: A schematic diagram showing the interactions of E-AHPA with neighboring groups within the active site of ODC (PDB code 2ON3)
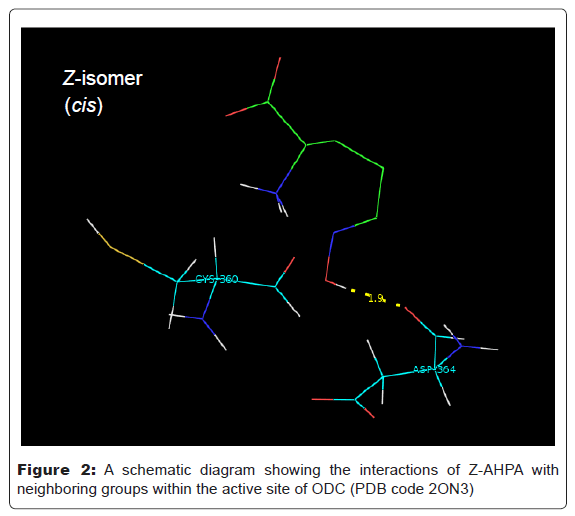
Figure 2: A schematic diagram showing the interactions of Z-AHPA with neighboring groups within the active site of ODC (PDB code 2ON3)
To evaluate the impact of ODC inhibitors on cell proliferation, MCF-7 cancer cells were used. The cells were cultured in the presence or absence of varying doses of AHPA. As shown in figure 3, AHPA treatment significantly reduced cell proliferation. A seven point dilution series was performed, and even at the lowest concentration (0.391 mg/ml), AHPA was found to affect cell viability. At 24 hours post-treatment, the cells were viable and the level of ATP was roughly equivalent for cells under each experimental condition. By 48 hours post-treatment, there was no proliferation detected and viability of all cells that had been exposed to AHPA was decreasing. In contrast, the control cells exhibited healthy and robust proliferation, as evidenced by the increasing amounts of ATP detected. By 96 hours, cells that had been exposed to AHPA at any dose were dead. It thus appears that AHPA was a much more potent inhibitor of cell proliferation than comparable doses of DFMO (difluoromethylornithine), a well-known inhibitor of ornithine decarboxylase. Although there is a possibility that the reduction of cell proliferation by AHPA is due to the compound toxicity, the significant effect of AHPA on cell proliferation and the in silico analysis support the conclusion that the AHPA inhibit the cell proliferation by binding to ODC.
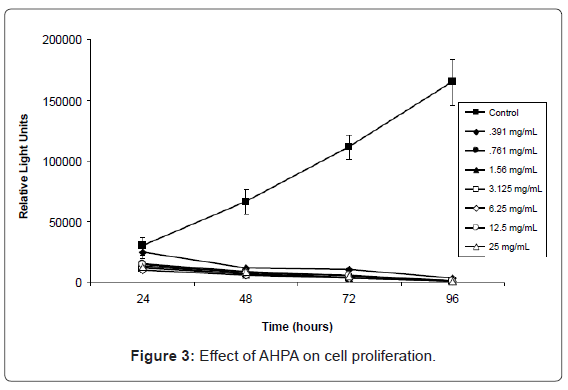
Figure 3: Effect of AHPA on cell proliferation.
`MCF-7 breast cancer cells were cultured in the presence of absence of AHPA at the indicated time points. Cell viability and proliferation was measured via the Cell Titer Glo Assay, and results are expressed as relative light units.
As shown in figure 4, cell proliferation was not significantly affected by doses of DFMO lower than 3 mg/ml. At doses higher than 3 mg/ml DFMO, modest reduction of cell proliferation was observed. Proliferation of cells treated with AHPA was dramatically reduced by even the lowest doses of the compound. These results demonstrate that AHPA is a potent inhibitor of cell proliferation.
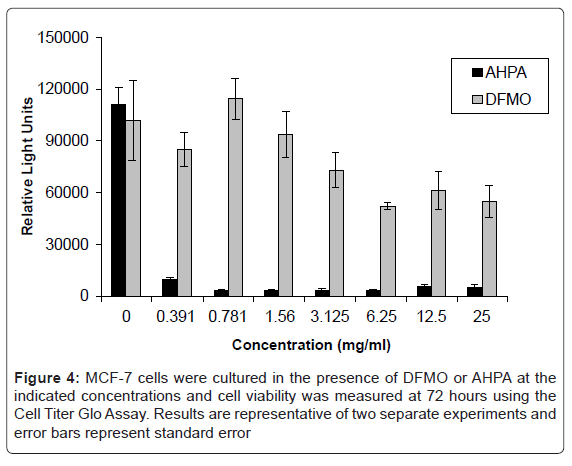
Figure 4: MCF-7 cells were cultured in the presence of DFMO or AHPA at the indicated concentrations and cell viability was measured at 72 hours using the Cell Titer Glo Assay. Results are representative of two separate experiments and error bars represent standard error
In conclusion, while exploring the design of new therapeutic inhibitors in arginine biosynthetic pathways, AHPA appears to be a potential cancer chemotherapeutic agent based on the observations of in silico docking and cell proliferation experiments. The docking studies have shown that geometric isomers of the ornithine-based oxime AHPA play a significant biological role in blocking ODC activity. The predicted binding affinity of the E-isomer of AHPA is higher than that of its Z-isomer with ODC as calculated with AutoDock. Besides, AHPA significantly affects cell viability with MCF-7. It is, however, worth noting that the in silico and cell proliferation observations will not warrant the fact that the AHPA specifically binds to ODC. To assess this issue, more evidence by further investigations would conclusively support the binding specificity. Furthermore, exploring structure-activity relationships of additional AHPA analogues as well as determining their lowest effective doses would be considered as future studies.
Experimental section
Synthesis of an amino acid oxime analogue: Reactions requiring anhydrous conditions were carried out under a nitrogen atmosphere in oven-dried glassware, and solvents were freshly distilled. Diethyl ether and Tetrahydrofuran (THF) were distilled from sodium/benzophenone. Dichloromethane and triethylamine were distilled from calcium hydride. All other reagents and solvents were used without further purification from commercial sources. Organic extracts were dried over anhydrous MgSO4 or Na2SO4. Reactions were monitored by Thin-Layer Chromatography (TLC) with 0.25-mm E. Merck precoated silica gel plates and visualized with ninhydrin solution (0.1% ninhydrin in 95% n-butanol, 4.5% water, 0.5% glacial acetic acid) and KMnO4 solution (3 g of KMnO4, 20 g of K2CO3, 5 ml of 5% NaOH, and 300 ml of water). Flash column chromatography was carried out with E. Merck silica gel 60 (230-240 mesh ASTM). The 1H and 13C NMR spectra taken by Dr. Garner by were recorded on a Bruker AM-500 spectrometer. Chemical shifts were expressed in parts per million (ppm) and referenced to CDCl3, CD3OD, or DMSO-d6 at Baylor University. The general syntheses of (S)-tert-butyl 2- (bis(tert-butoxycarbonyl)amino)-5-(hydroxyimino)pentanoate 5 and (S)-2-amino-5-(hydroxyimino)pentanoic acid (AHPA) 6 are as follows.
Procedure
(S)-tert-butyl2-(bis(tert-butoxycarbonyl)amino) -5- (hydroxyimino)pentanoate, 5
At room temperature to a solution of the appropriate aldehyde (1.0 g, 8.9 mmol) in methanol (5 ml) was added hydroxyamine hydrochloride (0.743 g, 0.107 mmol) followed by sodium acetate trihydrate (0.148 g, 0.107 mmol) and methanol (10 ml). Enough methanol was then added to give a clear solution, and stirring at 60°C was refluxed for 3-4 hrs. The resulting solution was cooled to room temperature, diluted with water (10 ml), and extracted with ether (3×10 ml). The combined organic phases were washed with saturated sodium bicarbonate (2×10 ml) and brine and then dried over MgSO4. The solvent was removed at reduced pressure, and the crude oximes was purified by column chromatography (1:9=ethylacetate:hexanes): clear oil. Yield (0.5 g 81.45%).
1H NMR (CDCl3): δ 7.49-7.41 (t, 0.56H), 6.83-6.73 (s, 0.47H), 4.85-4.73 (m, 1H), 2.15-1.99 (m, 1H), 1.54-1.40 (m, 29H). IR (cm-1): 3550-2800, 1800-1700. MS (ES+) (m/z): [M+Na] calcd for (C19H34N2O7Na) 425.2264, found 424.2245.
-amino-5-(hydroxyimino)pentanoic acid (AHPA) 6: To a stirred solution of the compound 5 (0.425 g, 1.03 mmol) in CH2Cl2 (10 ml) was slowly added Trifluoroacetic Acid (TFA) (8 ml) at 0°C. The reaction mixture was stirred for 1-4 hrs and then allowed to equilibrate at room temperature until the end of the reaction. The solvent was removed on a rotary evaporator, and the crude residue was purified by silica gel chromatography to yield 6 (0.24 g, 87% yield).
1H NMR (MeOD): δ 7.45-7.33 (m, 0.39H), 6.78-6.68(m, 0.29H), 4.08-3.90 (m, 1H), 2.25-1.95(m, 2H), δ1.38-1.20(m, 0.72H). IR (cm-1): 3500-2100, 1800-1600.
MS (ES+) (m/z): [M+H] calcd for (C5H11O3N2) 147.0770, found 147.0764.
The 3D structure of ODC (Ornithine Decarboxylase) from human and Leishmania donovani was obtained from the protein data bank (PDB code 2ON3). The X-ray crystal structure of the enzyme has been determined at 3.0 Å resolution [14]. All atom parameters were automatically created by AutoDockTools (ADT). Coordinates for the ODC (PDB code 2ON3) were processed in ADT by adding hydrogen atoms, assigning charges with the Gasteiger method, and merging the non-polar hydrogens. The 3D structure and active site residues of target protein were confirmed by PyMol [27].
To increase the validity of potential inhibitor and docking studies, inhibitor candidates (geometric isomers (E/Z) of oxime ligands) were chosen as control dockings. We utilized the Dundee PRODRG28 server to generate a topological description of the both E and Z form. This server converted 2D compounds drawn by the JME editor to 3D coordinates in PDB format, adding hydrogen atoms. The 3D structures of the compounds, in the PDB format, were then docked with the (PDB code 2ON3) using AutoDock4.0 (http://autodock.scripps.edu). A grid map of 60Å×60Å×60Å points with 0.375Å grid spacing was centered on the active site and was calculated around the docking area by running AutoGrid. The Lamarckian genetic algorithm was used as the search method, and the docking parameters were set to 10 automated docking runs, as default setup, for a 150 population size with a 2,500,000 maximum number of energy evaluations for each docking experiment. The results showed the binding energies and hydrogen bonds’ interactions between the ligands and (PDB code 2ON3), and the cluster analysis was performed based on Root Mean Square Deviation (RMSD).
MCF-7 breast cancer cells were maintained in FK12/DMEM medium supplemented with 5% fetal bovine serum in a 37°C incubator with a humidified 5% CO2 atmosphere. Cells were plated at 5×104 per well in white 96 well plates in the presence or absence of compound as indicated and each treatment was performed in triplicate. Cell growth was measured at the indicated time points using the Cell Titer Glo Kit (Promega, Madison, WI). Briefly, a luciferin substrate added to each well was converted to oxyluciferin in the presence of O2 and ATP. The light produced was measured using a Veritas Microplate Luminometer (Turner Biosystems, Sunnyvale, CA) and the relative light units were proportional to the amount of ATP present, reflecting the number of viable cells in the well. DFMO was purchased from Sigma-Aldrich (St. Loius, MO).
We thank Professor Charles Garner for data analysis of 1H, 13C NMR supported by the NSF (Award #CHE-042802) and Dr. Alejandro Ramirez (High Resolution Mass Spectroscopy) at Baylor University. We acknowledge the Robert A. Welch Foundation to Department of Chemistry and Biochemistry at McMurry University and Lily Drake Cancer Research.