Journal of Theoretical & Computational Science
Open Access
ISSN: 2376-130X
ISSN: 2376-130X
Research Article - (2016) Volume 3, Issue 2
The 2-hydroxy-5-(Phenyldiazenyl)benzaldehyde oxime (PDBO) was synthesized and characterized. The spectral investigations such as FT-IR, FT-Raman and UV-Vis spectra were recorded. The bond parameter values were calculated at DFT/B3LYP/6- 311++G(d,p) level of theory. The observed spectral results were compared with the computed wavenumber. The complete vibrational assignments of wavenumbers were made on the basis of TED. The first order hyperpolarizability, intra-molecular charge transfer and band gap energy were studied using B3LYP/6-311++G(d,p) calculation. The electronic transition was studied using UV-Vis spectrum and the observed values were compared with the theoretical values. The MEP, Mulliken charges and thermodynamic parameters of the title molecule was also analyzed using the same level of basis set.
<Keywords: FT-IR; FT-Raman; TED; NBO; PDBO
The Azo compounds are most versatile molecule and it has received much attention in research in the view of both fundamental and its applications [1,2]. In recent year a large amount of investigation has been carried out for the synthesis and spectroscopic properties of this group of dyes [3-5]. Furthermore, azo compounds are known to be involved in a number of biological reactions such as inhibition of DNA, RNA, and protein synthesis, carcinogenesis, and biological activity against bacteria and fungi [6,7]. The azo dyes produced by aromatic and their oxime derivatives containing at least one azo group (-N=N-) attached to at least one aromatic moiety are the most widely used dyes in textile, printing, leather, paper making, drug and food industries [8]. The azo compounds are very attractive from theoretical and practical viewpoints and have been of particular interest [9-11]. It is described by the intra-molecular charge transfer between the phenol and oxime groups in ground or excited state [12].
Aromatic and heteroaromatic azo compounds constitute the largest and the most diverse group of synthetic dyes with application not only as textile colorants but in many other industrial fields for coloring different substrates, biological-medical studies, in the field of non-linear optics and optical data storage [13-15]. The interest for benzothiazole azo dyes as disperse and cationic dyes for textile application is growing [16]. In recent years, with the development of density functional theory and especially the improvement of time-dependent density functional theory [17], properties of both ground- and excited-states for mediumsized metal complexes can be calculated at the first-principle level with good accuracy [18,19]. To get better insight into the geometry and the electronic structure geometry optimizations of the ground and excitedstates were carried out by means of DFT calculations. We also calculate and analyze the three excited states derived from TD-DFT results and compare them with the ground state molecular orbitals (MOs) obtained by DFT calculations.
The entire calculations were performed at DFT/B3LYP/6- 311++G(d,p) level of basis set using Gaussian 03W [20] program package, invoking gradient geometry optimization [20,21]. The optimized structural parameters were used in the vibrational frequency calculations at the DFT level to characterize all the stationary points as minima. The vibrationally averaged nuclear positions of PDBO were used for harmonic vibrational frequency calculations resulting in IR and Raman frequencies together with intensities and Raman depolarization ratios. The vibrational modes were assigned on the basis of TED analysis using VEDA4 program [22]. The Raman activity was calculated by using Gaussian 03W package and the activity was transformed into Raman intensity using Raint program [23] by the expression:
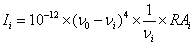
Where Ii is the Raman intensity, RAi is the Raman scattering activities, νi is the wavenumber of the normal modes and ν0 denotes the wavenumber of the excitation laser [24].
Synthesis procedure.
A mixture of 5-arylazosalicylaldehyde (1 mmol) and sodium acetate (0.5 g) was dissolved in boiling ethanol and hydroxylamine hydrochloride (0.13 g; 2 mmol) was added. The mixture was refluxed for 3 hr. The reaction mixture was poured into water. The 5-arylazo salicylaldoxime separated out was filtered and recrystallized from ethanol.
Molecular geometry
The geometrical parameters such as bond distance (Å), bond angles (˚) and torsional angles (˚) of PDBO were calculated using B3LYP method with 6-311++G (d,p) basis set. The computed values along with the available experimental data [25] are presented in Table 1 and the optimized structure of PDBO are also shown in Figure 1. The molecule PDBO has two rings namely: i) benzene and ii) phenonilc oxime and the azo chromophore group (-N=N-) is connected in between them. As it is evident from the Table 1, both the Azo benzene rings are coplanar: C2-C3-N12-N13=-180.00 and C4-C3-N12-N13=0.00 and C16-C14-N13-N12=0.00 and C15-C14-N13-N12=-180.00°. The bond angles between C-N and benzene rings are equal: C2-C3-N12=C15- C14-C13=115˚ and C4-C3-N12=C16-C14-N13=124˚ which show there is no steric repulsion in between Azo and benzene rings. In this study, the N=N bond length is 1.253 Å which is supported by literature value 1.25 Å [26,27]. The bond lengths of C3-N12 and C14-N13 are differ by 0.002 Å, which is due to the energy transfer takes place during π(N12=N13) to π*(C2-C3/C14-C15) transitions are differ by E(2) value: 0.01 kj/mol (Table 4). All the calculated C-C and C-H bond distances are in agreement with our earlier work [28] with few exceptions.
| Parameters | B3LYP/6-311++G(d,p) | XRD [25] |
|---|---|---|
| Bond Lengths (Å) | ||
| C3-N12 | 1.417 | 1.428 (9) |
| N12-N13 | 1.252 | 1.257 (9) |
| N13-C14 | 1.415 | |
| C16-C19 | 1.395 | 1.39 |
| C21-O23 | 1.367 | 1.367 (7) |
| O23-H24 | 0.962 | 0.84 |
| C25-H26 | 1.093 | |
| C25-N27 | 1.277 | 1.304 (14) |
| N27-O28 | 1.379 | |
| O28-H29 | 0.974 | |
| Bond Angles (°) | ||
| C2-C3-N12 | 115.3 | |
| C4-C3-N12 | 124.7 | |
| C3-N12-N13 | 115.5 | 113.4 (7) |
| N12-N13-C14 | 115.4 | 112.2 (7) |
| N13-C14-C15 | 115.5 | |
| N13-C14-C16 | 124.8 | |
| C16-C19-C21 | 118.5 | 120 |
| C21-C19-C25 | 119.2 | 121.2 (5) |
| C21-O23-H24 | 109.9 | 109.5 |
| Dihedral Angles (°) | ||
| C2-C3-N12-N13 | -180.0 | |
| C4-C3-N12-N13 | -0.001 | |
| C3-N12-N13-C14 | -179.9 | |
| N12-N13-C14-C15 | -180.0 | |
| N12-N13-C14-C16 | -0.008 | |
| N13-C14-C15-C17 | 179.9 |
Table 1: The selected bond parameters of PDBO.

Figure 1: The optimized structure of 2-hydroxy-5-(Phenyldiazenyl) benzaldehyde oxime (PDBO).
Vibrational assignments
The compound PDBO belongs to C1 point group symmetry and it consists of 29 atoms. Hence 81 normal modes of vibrations are possible and are distributed as: Γ55A′ planar+26″ non-planar). The vibrational wavenumbers and their corresponding assignments are given in Table 2. The vibrational assignments are made on the basis of the total energy distribution (TED) calculations. The theoretical and observed (FT-IR and FT-Raman) spectra are shown in Figures 2 and 3.
| Mode No | Calculated Frequencies (cm-1) | Observed Frequencies (cm-1) | IR Intensity | Raman Intensity | Reduced Masses | Force Consts | Vibrational Assignments≥10% (TED)d | |
|---|---|---|---|---|---|---|---|---|
| Scaleda | FT-IR | FT-Raman | Rel.b | Rel.c | ||||
| 1 | 3685 | 3854 | 45.53 | 0.62 | 1.07 | 9.24 | ѴO23H24(100) | |
| 2 | 3480 | 3416 | 0.79 | 0.91 | 1.07 | 8.24 | ѴO28H29(100) | |
| 3 | 3095 | 1.39 | 0.04 | 1.09 | 6.66 | ѴC16H20(99) | ||
| 4 | 3084 | 1.63 | 0.24 | 1.09 | 6.63 | ѴC4H9(92) | ||
| 5 | 3070 | 0.14 | 0.76 | 1.09 | 6.58 | ѴC1H7(92) | ||
| 6 | 3070 | 5.64 | 0.44 | 1.10 | 6.59 | ѴC15H18(82) | ||
| 7 | 3061 | 3064 | 3065 | 6.64 | 0.89 | 1.09 | 6.53 | ѴC1H7(85) |
| 8 | 3050 | 3.67 | 0.62 | 1.09 | 6.47 | ѴC2H8(88) | ||
| 9 | 3040 | 3040 | 0.84 | 0.19 | 1.09 | 6.40 | ѴC1H7(84) | |
| 10 | 3033 | 6.95 | 0.71 | 1.09 | 6.40 | ѴC17H22(45)+ѴC25H26(41) | ||
| 11 | 2945 | 2923 | 15.63 | 0.47 | 1.09 | 6.02 | ѴC17H22(99) | |
| 12 | 1603 | 1622 | 1622 | 26.05 | 8.48 | 7.68 | 12.61 | ѴN27C25(76) |
| 13 | 1575 | 1575 | 1591 | 2.42 | 6.19 | 6.38 | 10.10 | ѴN12N13(10)+ѴC1C2(47) |
| 14 | 1569 | 15.46 | 5.81 | 6.42 | 10.08 | ѴC1C2(48)+ѴC19C25(13) | ||
| 15 | 1562 | 9.07 | 0.70 | 6.52 | 10.16 | ѴC2C3(39)+βC14C16C19(11) | ||
| 16 | 1557 | 5.58 | 0.47 | 5.87 | 9.08 | ѴC1C2(36)+ѴC1C2(10)+βC1C2C3(12) | ||
| 17 | 1483 | 1482 | 1493 | 9.08 | 100.00 | 6.52 | 9.15 | ѴN12N13(63) |
| 18 | 1462 | 1466 | 26.94 | 0.17 | 2.87 | 3.92 | βH20C16C19(45) | |
| 19 | 1447 | 1437 | 3.61 | 25.25 | 2.34 | 3.13 | ѴC1C2(12)+βH8C2C1(38) | |
| 20 | 1421 | 2.20 | 18.55 | 2.38 | 3.07 | βH7C1C6(48) | ||
| 21 | 1396 | 1394 | 1377 | 17.04 | 41.49 | 4.24 | 5.27 | ѴC1C2(48) |
| 22 | 1390 | 14.42 | 1.74 | 1.52 | 1.87 | βH29O28N27(47)+βH26C25N27(23) | ||
| 23 | 1311 | 1301 | 1312 | 5.38 | 2.27 | 4.08 | 4.48 | ѴC14C16(66)+βH24O23C21(14) |
| 24 | 1296 | 0.92 | 0.55 | 4.04 | 4.32 | ѴC1C2(64)+ βH7C1C6(13) | ||
| 25 | 1283 | 2.56 | 5.94 | 1.42 | 1.49 | βH29O28N27(12)+βH7C1C6(49)+βH26C25N27(10) | ||
| 26 | 1282 | 1.06 | 0.71 | 1.42 | 1.49 | βH29O28N27(22)+βH8C2C1(36) | ||
| 27 | 1254 | 1266 | 7.38 | 0.83 | 1.74 | 1.75 | ѴN12C3(11)+βH18C15C17(49) | |
| 28 | 1236 | 1248 | 1247 | 100.00 | 10.65 | 4.52 | 4.41 | ѴC15C17(62) |
| 29 | 1192 | 1195 | 1195 | 15.18 | 0.09 | 2.23 | 2.02 | ѴN12C3(18) |
| 30 | 1171 | 9.69 | 38.71 | 2.09 | 1.83 | ѴC16C19(14)+ѴN13C14(12)+βH24O23C21(15) | ||
| 31 | 1135 | 1142 | 1144 | 0.61 | 0.84 | 1.15 | 0.94 | βH7C1C6(61) |
| 32 | 1133 | 8.71 | 0.13 | 1.27 | 1.04 | βH24O23C21(23)+βH18C15C17(39) | ||
| 33 | 1131 | 3.00 | 1.16 | 1.37 | 1.12 | βH9C4C5(40) | ||
| 34 | 1113 | 1107 | 1108 | 7.42 | 83.07 | 2.26 | 1.79 | ѴN12C3(27)+βH9C4C5(10) |
| 35 | 1065 | 1070 | 45.21 | 18.06 | 1.92 | 1.39 | ѴC19C25(17)+βH18C15C17(23) | |
| 36 | 1054 | 1014 | 3.89 | 2.28 | 1.51 | 1.07 | ѴC5C6(31)+βH7C1C6(11)+βH7C1C6(24) | |
| 37 | 998 | 999 | 3.29 | 0.59 | 2.05 | 1.30 | ѴC1C6(46)+βH10C5C6(24)+βC4C5C6(14) | |
| 38 | 979 | 16.80 | 0.41 | 2.82 | 1.72 | βH20C16C19(10)+βC3N12N13(10)+βC14C16C19(10) | ||
| 39 | 975 | 0.29 | 9.18 | 6.15 | 3.73 | ѴC1C6(25)+βC1C6C5(20)+βC2C1C6(43) | ||
| 40 | 964 | 0.03 | 0.04 | 1.29 | 0.77 | τH7C1C6C5(62)+τH2C1C6C5(19) | ||
| 41 | 957 | 956 | 64.86 | 0.66 | 8.20 | 4.79 | ѴO28N29(67) | |
| 42 | 953 | 0.00 | 0.00 | 1.36 | 0.79 | τH9C4C5C6(82) | ||
| 43 | 934 | 6.87 | 0.10 | 1.48 | 0.82 | τH20C16C19C21(77) | ||
| 44 | 913 | 1.56 | 0.12 | 1.37 | 0.73 | τH18C15C17C21(80) | ||
| 45 | 910 | 1.66 | 0.02 | 1.45 | 0.77 | τH11C6C5C4(79) | ||
| 46 | 905 | 899 | 0.01 | 0.03 | 1.39 | 0.72 | τH20C16C19C21(72) | |
| 47 | 876 | 1.50 | 0.06 | 6.33 | 3.10 | βC14C15C17(24)+βC2C1C6(13) | ||
| 48 | 820 | 832 | 0.02 | 0.00 | 1.25 | 0.54 | τH7C1C6C5(96) | |
| 49 | 809 | 7.12 | 1.82 | 5.76 | 2.41 | ѴC15C17(33)+βC15C14C16(18) | ||
| 50 | 785 | 789 | 789 | 16.88 | 0.02 | 1.51 | 0.60 | τH18C15C17C21(69) |
| 51 | 770 | 3.82 | 0.78 | 6.22 | 2.36 | ѴN12C3(13)+βC1C6C5(21)+βC14C15C17(11) | ||
| 52 | 754 | 759 | 9.07 | 0.08 | 1.78 | 0.65 | τH9C4C5C6(32)+τH7C1C6C5(16)+ГN12C2C4C3(18) | |
| 53 | 709 | 2.30 | 0.15 | 4.15 | 1.33 | τC3C2C1C6(12)+ГC25C16C21C19(50) | ||
| 54 | 672 | 682 | 14.26 | 0.00 | 1.90 | 0.55 | τH9C4C5C6(50)+τC2C1C6C5(15)+ГC17C21O23H24(25) | |
| 55 | 635 | 2.47 | 0.82 | 6.15 | 1.58 | βC14C16C19(18)+βC14C15C17(16)+βC25N27O28(33) | ||
| 56 | 633 | 0.10 | 0.80 | 6.98 | 1.78 | βC3N12N13(42) | ||
| 57 | 604 | 606 | 613 | 0.07 | 1.18 | 6.53 | 1.52 | βC2C1C6(72) |
| 58 | 592 | 598 | 1.66 | 0.01 | 3.62 | 0.81 | τC2C1C6C5(11)+τC3C2C1C6(34)+τC14N13N12C3(11) | |
| 59 | 549 | 550 | 0.97 | 0.09 | 4.65 | 0.89 | βC3N12N13(58)+ГC19C21O23H24(20) | |
| 60 | 501 | 506 | 3.55 | 0.00 | 3.32 | 0.53 | τC2C1C6C5(14)+τC3C2C1C6(11)+ГN12C2C4C5(31) | |
| 61 | 496 | 3.70 | 0.07 | 7.24 | 1.14 | ѴN15C17(14)+βC15C14C16(18) | ||
| 62 | 469 | 476 | 16.13 | 0.16 | 7.38 | 1.04 | βC14N13N12(15)+βC14C16C19(34) | |
| 63 | 459 | 454 | 5.39 | 0.00 | 3.13 | 0.42 | τC19C25N27O28(68) | |
| 64 | 429 | 423 | 432 | 22.46 | 0.33 | 1.28 | 0.15 | τH29O28N27C25(91) |
| 65 | 403 | 0.00 | 0.00 | 2.96 | 0.31 | τC1C6C5C4(78) | ||
| 66 | 392 | 0.02 | 0.26 | 5.35 | 0.53 | τC3C2C1C6(10)+τC14C15C17C21(54) | ||
| 67 | 391 | 13.52 | 0.09 | 5.73 | 0.56 | βC3N12N13(11)+βC3N12N13(57) | ||
| 68 | 322 | 2.50 | 0.92 | 6.95 | 0.46 | βC14C16C19(25) | ||
| 69 | 305 | 19.44 | 0.18 | 1.85 | 0.11 | τH24O23C21C17(57)+τH24O23C21C17(15) | ||
| 70 | 293 | 14.61 | 0.33 | 1.68 | 0.09 | τH3C2C1C6(12)+τH24O23C21C17(59) | ||
| 71 | 267 | 273 | 0.29 | 0.53 | 5.69 | 0.26 | ѴN13C14(11)+βC15C14C16(10)+βC14N13N12(35) | |
| 72 | 257 | 0.00 | 0.40 | 5.79 | 0.24 | τC16C14C15C17(50) | ||
| 73 | 200 | 1.22 | 0.05 | 6.55 | 0.17 | ѴN13C14(11)+βC3C12C13(44) | ||
| 74 | 182 | 177 | 0.01 | 0.09 | 8.17 | 0.17 | τC2C3N12N13(22)+τC15C17C21C19(36) | |
| 75 | 122 | 9.09 | 1.79 | 3.46 | 0.03 | ГC16C15N13C14(68) | ||
| 76 | 119 | 1.57 | 1.33 | 6.66 | 0.06 | βC3N12N13(75) | ||
| 77 | 91 | 3.64 | 0.08 | 5.11 | 0.03 | τC21C19C25N27(12)+τC2C3N12N13(46)+τC2C1C6C5(11) | ||
| 78 | 66 | 70 | 5.55 | 2.58 | 3.64 | 0.01 | τC2C1C6N5(79) | |
| 79 | 51 | 0.59 | 5.36 | 6.56 | 0.01 | βC14N13N12(79) | ||
| 80 | 47 | 2.01 | 0.02 | 6.09 | 0.01 | τC14N13N12C3(39)+τC14C15C17C21(19)+ГN12C2C4C3(14) | ||
| 81 | 16 | 0.09 | 8.09 | 4.05 | 0.00 | τC2C3N12N13(87) | ||
aScaling factor: 0.9608 ,bRelative IR absorption intensities normalized with highest peak absorption equal to 100,c Relative Raman intensities calculated by Equation (1) and normalized to 100. dTED calculated at B3LYP/6-311++G(d,p) level.
Table 2: The vibrational assignments of PDBO.

Figure 2: Theoretical and Experimental FT-IR spectra of PDBO.

Figure 3: Theoretical and Experimental FT-Raman spectra of PDBO.
The calculated frequencies are slightly higher than the observed values for the majority of the normal modes. Two factors may be responsible for the discrepancies between the experimental and computed spectra of azo compound. The first is caused by the environment and the second reason for these discrepancies is the fact that the experimental value is an enharmonic frequency while the calculated value is a harmonic frequency [29].
O-H vibrations: The OH group vibrations are likely to be most sensitive to the environment, so they show pronounced shifts in the spectra of the hydrogen-bonded species. The non-hydrogen-bonded or a free hydroxyl group absorb strongly in the 3550-3700 cm−1 region [30]. In the present study, the calculated/observed OH stretching vibrations are 3685, 3480/3854, 3416 cm−1 (mode nos: 1, 2). The phenolic νO23H24 vibration observed at higher frequency (~438 cm-1) than the oxime hydroxy (νO29H30) mode, which is due to the large ionic character of the phenolic OH group.
The OH in-plane bending vibration in phenol, lies in the region 1270-1150 cm−1 [31] and is not much affected due to hydrogen bonding. In our case, the observed FT-IR/FT-Raman bands appeared at 1301/1312 cm−1 and its corresponding calculated wavenumber 1311 cm-1 (mode no: 23) with TED 14% are belong to βOH mode. The OH out-of-plane deformation vibration in phenol lies in the region 517- 710 cm−1 for associated OH [32]. In this study, the OH out-of-plane bending vibrations observed at 682 cm-1 in FT-IR spectrum and its calculated value is 672 cm-1 (mode no: 54). In both inter-molecular and intra-molecular associations, the frequency is at a higher value than in free OH [33]. The frequency increases with hydrogen bond strength because of the larger amount of energy required to twist the O-H bond for out-of plane bending [34]. The mode nos: 26 and 64 are assigned to βO28H29 and ΓO28H29 modes, respectively.
C-H Vibrations: The hetero aromatic structure shows the presence of C-H stretching vibrations in the region 3200-2900 cm−1 [31,35], which is the characteristic region for the ready identification of the C-H stretching vibrations and in this region, the bands are not affected appreciably by the nature of the substituents. In this study, the molecule PDBO have nine νCH vibrations, which are assigned in the region 3084- 2945 cm-1 (mode nos: 4 to 11). This assignment is further supported by observed FTIR (3064, 2923 cm-1) and FT-Raman bands (3065, 3040 cm-1) and also find support from literature [33]. Literature survey reveals that the CH in-plane/out-of-plane bending vibrations appear in the ranges 1000-1300 cm-1/750-1000 cm-1, respectively [36,37]. Based on the fact, the harmonic wavenumbers in the regions 1462-1014 cm-1 (mode nos: 18-20, 25-27, 31-33, 36)/964-672 cm-1 (mode nos: 40, 42-46, 48, 50, 52, 54) are assigned to CH in-plane/out-of-plane bending modes, respectively. These assignments find support from the observed spectral values: βCH (FTIR: 1014, 1142, 1266 cm-1)/FT-Raman: 1144, 1437, 1466 cm-1) and ΓCH (FTIR: 899, 832, 789, 759, 682 cm-1)/FT-Raman: 789 cm-1) and also in line with literature [28].
C-C vibrations: The benzene ring modes predominantly involve C-C bonds and the vibrational frequency associated with C-C stretching modes of carbon skeleton. In general, the C-C stretching modes, known as semi-circle stretching, modes observed in the range of 1510- 1660 cm-1 [31]. The phenyl ring C-C stretching vibrations occur in the regions of 1625-1590, 1590, 1575, 1465-1430 and 1380-1280 cm-1 were given by Varsanyi [32]. The harmonic wavenumbers in the range 1575- 998 cm-1 mode nos: 13-16, 19, 21, 23, 24, 28, 30, 36, 37) are assigned to νC-C modes. The observed FTIR: 1575, 1394, 1301, 1248 and 1014 cm-1 and FT-Raman: 1591, 1437, 1377, 1312, 1247 and 999 cm-1 bands support the νC-C mode. In which the harmonic (1311/mode no: 23)/ observed bands 1301: FTIR/1312 cm-1: FT-Raman) are relatively at a higher wavenumber due to its coupling with βO23H24 bending mode [33]. The coupling C19-C25 stretching vibration computed at 1065 cm-1 (mode no: 35) show good agreement with recorded FTIR band at 1070 cm-1. In this study, the harmonic C-C-C in-plane and out-of-plane bending modes have been found to be in agreement with the observed spectral values.
(-C-N=N-C-) vibrations: In this study, the Azo chromophoric group can be considered as a single unit, which contains two bonds: C-N and N=N. Each vibrations has its own well known characteristic vibrational frequency. In PDBO the two unequivalent C-N parts C3-N12: 1.4178 Å and C14-N13: 1.4159 Å causes a change in dipolemoment and hence the N=N stretching is IR active with medium strong intensity (1482 cm-1: FTIR), while the corresponding Raman bands is 1493 cm-1 (weak). The N=N stretching vibration of Azo compounds show bands in the region 1380-1450 cm-1 [38]. Literature survey reveals, that the bands observed between 1200-1300 cm-1 [39,40] belongs to the νC-N modes of Azo compound. The observed FTIR/FT-Raman bands 1107/1108 cm-1 and harmonic frequency 1171 cm-1 (mode no: 30) and are assigned to νC3-N12 and νC14-N13 modes, respectively. These observations indicate that the C-N parts (C3-N12, C14-N13) of azo group have got increased double bond character [35].
Nonlinear optical properties
NLO materials have been the subject of intense research, due to their possible application in a wide range of technologies, such as optical communication, optical computing and data storage [41-43]. Therefore, it is known that there has been an intense investigation for molecules with large non-zero hyperpolarizabilities, since these substances have potential as the constituents of NLO materials. The azo compound containing systems have a special significance among the many molecular designs that are used for introducing NLO behavior [44,45]. Due to their peculiar photo switching properties, azo benzenes are used in many areas of molecular electronics and suitable for various kinds of applications [46-48].
In this study, the electronic dipole moment, molecular polarizability, anisotropy of polarizability and molecular first hyperpolarizability of PDBO were calculated at B3LYP/6-311++G(d,p) basis set. The mathematical details used for the calculations were reported in our earlier publications [28], and current results are presented in Table 3. It is wellknown that the higher values of dipole moment, molecular polarizability, and hyperpolarizability which enhances the NLO property.
| Parameters | B3LYP/6-311++G(d,p) |
|---|---|
| Dipole moment (μ)Debye | |
| μx | -0.708 |
| μy | -1.580 |
| μz | -0.0001 |
| μ | 1.731Debye |
| Hyperpolarizability (β0)×10-30esu | |
| βxxx | 1864.227 |
| βxxy | 180.539 |
| βxyy | -19.196 |
| βyyy | -322.478 |
| βxxz | 0.170 |
| βxyz | 0.036 |
| βyyz | 0.019 |
| βxzz | 106.305 |
| βyzz | -41.978 |
| βzzz | 0.003 |
| β0 | 16.932×10-30esu |
Table 3: The NLO measurements of PDBO.
Urea is one of the prototypical molecule used in the study of the NLO property of molecular systems, and thus, it was used frequently as a threshold value for comparative purposes. The first hyperpolarizability value of PDBO was calculated as 16.93289 × 10-30 esu using B3LYP/6- 311++G(d,p) method. According to these result, the β0 value of present molecule is 45 times larger than the magnitude of urea.
NBO analysis
The NBO analysis was performed on PDBO using B3LYP method with 6-311++G(d,p) basis set and the results are listed in the Table 4. NBO analysis provides an efficient method for studying intraand inter-molecular bonding and interaction among bonds, and also provides a convenient basis for investigating charge transfer or conjugative interaction in molecular systems. Some electron donor orbital, acceptor orbital and the interacting stabilization energy resulted from the second-order micro-disturbance theory are reported [49,50]. The larger E(2) value the more intensive is the interaction between electron donors and acceptor i.e., the more donation tendency from electron donors to electron acceptors and the greater the extent of conjugation of the whole system [51]. Delocalization of electron density between occupied Lewis - type (bond or lone pair) NBOs and formally unoccupied (anti bond or Rydeberg) non Lewis NBOs correspond to a stabilizing donor-acceptor interaction.
| Type | Donor NBO (i) | ED/e | Acceptor NBO (j) | ED/e | E(2) kj/mol |
E(j)-E(i) a.u. |
F(i,j) a.u. |
|---|---|---|---|---|---|---|---|
| σ -σ* | BD (1) C2 - C3 | 1.9753 | BD*(1) C1 - C2 | 0.01416 | 10.6 | 1.28 | 0.051 |
| BD*(1) C1 - H7 | 0.01379 | 9.7 | 1.14 | 0.046 | |||
| BD*(1) C3 - C4 | 0.03365 | 15.8 | 1.26 | 0.062 | |||
| BD*(1) C4 - H9 | 0.01564 | 9.0 | 1.15 | 0.045 | |||
| BD*(1) N12 - N13 | 0.00763 | 8.4 | 1.28 | 0.045 | |||
| π -π* | BD (2) C2 - C3 | 1.6128 | BD*(2) C1 - C6 | 0.32408 | 80.4 | 0.28 | 0.067 |
| BD*(2) C4 - C5 | 0.28956 | 80.8 | 0.28 | 0.068 | |||
| BD*(2) N12 - N13 | 0.21273 | 86.1 | 0.23 | 0.064 | |||
| σ -σ* | BD (1) C3 - N12 | 1.9794 | BD*(1) C1 - C2 | 0.01416 | 7.4 | 1.35 | 0.044 |
| BD*(1) C2 - C3 | 0.02184 | 4.1 | 1.33 | 0.033 | |||
| BD*(1) C3 - C4 | 0.03365 | 6.1 | 1.33 | 0.039 | |||
| BD*(1) C4 - C5 | 0.01452 | 4.8 | 1.36 | 0.035 | |||
| BD*(1) N13 - C14 | 0.03018 | 19 | 1.18 | 0.066 | |||
| σ -σ* | BD (1) N12 - N13 | 1.9868 | BD*(1) C2 - C3 | 0.02184 | 7.1 | 1.54 | 0.046 |
| BD*(1) C14 - C15 | 0.02186 | 7.4 | 1.52 | 0.046 | |||
| π -π* | BD (2) N12 - N13 | 1.9143 | BD*(2) C2 - C3 | 0.36971 | 42.4 | 0.4 | 0.061 |
| BD*(2) C14 - C15 | 0.39365 | 42.1 | 0.39 | 0.061 | |||
| σ -σ* | BD (1) N13 - C14 | 1.9793 | BD*(1) C3 - N12 | 0.03041 | 18.8 | 1.19 | 0.065 |
| BD*(1) C14 - C15 | 0.02186 | 4.4 | 1.33 | 0.034 | |||
| BD*(1) C14 - C16 | 0.03187 | 7.1 | 1.34 | 0.043 | |||
| BD*(1) C15 - C17 | 0.01286 | 7.2 | 1.34 | 0.043 | |||
| BD*(1) C16 - C19 | 0.0192 | 5.0 | 1.34 | 0.036 | |||
| π -π* | BD (2) C16 - C19 | 1.6515 | BD*(2) C14 - C15 | 0.39365 | 72.2 | 0.28 | 0.063 |
| BD*(2) C17 - C21 | 0.38858 | 103.2 | 0.27 | 0.073 | |||
| BD*(2) C25 - N27 | 0.16239 | 67.9 | 0.27 | 0.062 | |||
| σ -σ* | BD (1) C17 - C21 | 1.978 | BD*(1) C15 - C17 | 0.01286 | 12.8 | 1.3 | 0.057 |
| BD*(1) C15 - H18 | 0.01333 | 9.58 | 1.17 | 0.046 | |||
| BD*(1) C19 - C21 | 0.03049 | 18.2 | 1.26 | 0.066 | |||
| BD*(1) C19 - C25 | 0.02776 | 12.0 | 1.19 | 0.052 | |||
| π -π* | BD (2) C17 - C21 | 1.6503 | BD*(2) C14 - C15 | 0.39365 | 96.3 | 0.3 | 0.075 |
| BD*(2) C16 - C19 | 0.33865 | 61.9 | 0.3 | 0.06 | |||
| σ -σ* | BD (1) O23 - H24 | 1.9878 | BD*(1) C19 - C21 | 0.03049 | 17.2 | 1.3 | 0.065 |
| σ -σ* | BD (1) C25 - H26 | 1.981 | BD*(1) C16 - C19 | 0.0192 | 16.2 | 1.13 | 0.059 |
| BD*(1) N27 - O28 | 0.02116 | 6.4 | 0.79 | 0.031 | |||
| σ -σ* | BD (1) C25 - N27 | 1.9917 | BD*(1) C19 - C21 | 0.03049 | 5.4 | 1.45 | 0.039 |
| BD*(1) C19 - C25 | 0.02776 | 8.5 | 1.38 | 0.048 | |||
| π -π* | BD (2) C25 - N27 | 1.9519 | BD*(2) C16 - C19 | 0.33865 | 31.3 | 0.37 | 0.051 |
| σ -σ* | BD (1) N27 - O28 | 1.9886 | BD*(1) C19 - C25 | 0.02776 | 12.8 | 1.32 | 0.057 |
| σ -σ* | BD (1) O28 - H29 | 1.9937 | BD*(1) N27 - O28 | 0.02116 | 5.4 | 0.99 | 0.032 |
| n -σ* | LP (1) O23 | 1.978 | BD*(1) C17 - C21 | 0.02449 | 23.9 | 1.17 | 0.073 |
| n -π* | LP (2) O23 | 1.872 | BD*(2) C17 - C21 | 0.38858 | 116.1 | 0.36 | 0.095 |
Table 4: The NBO analysis for PDBO.
The lower electron density of donor and higher electron density of acceptor have maximum delocalization, which results strong bond interaction. In general, the bonds having higher electron density value with lower E(2) energy which causes the vibrational frequency shifts. The electron density E(2) energy value for C3-N12 and C14-N13 are 1. 9794 e/4.18 kj/mol and 1.9793 e/7.15 kj/mol, respectively and their corresponding harmonic values are 1113 and 1171 cm-1 mode nos: 34 and 30), which are negatively deviated from literature (1200-1300 cm-1) [39,40]. The LP O23-π transfer more energy to acceptor bond π* C17-C21 (116.11 kj/mol), while the LPO23-π transfer lesser energy to C25-N27 bond (84.52 kj/mol). This may be one of the reason for the phenolic O23-H24 stretching observed at higher frequency 3854 cm-1 than the oxime hydroxy OH νO28-H29: 3416 cm-1.
In this study the bonds σN12-N13 and πN12-N13 having the electron density 1.9868 e and 1.9143 e, which stabilizes 7.11 and 42.43 kj/mol, respectively to its antibonding orbitals of σ*(C2-C3) and π*(C2-C3). The more energy transfer takes place during π to π* transition rather than σ to σ*. The maximum stabilization energy E(2) associated with resonance interaction π(C16-C19)→π*(C17-C21) is obtained as 103.22 kj/mol, which is due to the O23-H24 group attached with C21 atom.
UV-Visible analysis
The UV-Vis absorption spectra of PDBO molecule were calculated using TD-DFT method with B3LYP/6-311++G(d,p) basis set. The observed and calculated UV-Visible spectra are shown in Figure 4 and their values are presented in Table 5.
| Calculated atB3LYP/6-311++G(d,p) | Oscillator strength | Calculated Band gap (ev/nm) |
Experimental Band gap (nm) |
Assignments |
|---|---|---|---|---|
| Excited State 1 | Singlet-A (f=0.0000) | 2.5897 eV/478.75 nm | 430 | π-π* |
| 62 -> 64 | 0.66567 | 3.953529 | ||
| Excited State 2 | Singlet-A (f=0.5172) | 3.4096 eV/363.64 nm | 350 | π-π* |
| 61 -> 64 | 0.17914 | 4.328773 | ||
| 63 -> 64 | 0.60836 | 3.760057 | ||
| 63 -> 65 | 0.18689 | 4.506191 | ||
| Excited State 3 | Singlet-A (f=0.2955) | 3.7765 eV/328.30 nm | 280 | π-π* |
| 61 -> 64 | 0.60751 | 4.328773 | ||
| 63 -> 64 | -0.18306 | -3.760057 | ||
| 63 -> 65 | 0.18127 | 4.506191 |
Table 5: The electronic transitions of PDBO.

Figure 4: Theoretical and Experimental UV-Visible spectra of PDBO.
The absolute errors between the theoretical and experimental emission λmax values decreases evidently when solvent effects are considered with the TD-DFT method. When the title compound is excited at 329, 363 and 478 nm in calculated spectrum, on the other hand the experimental spectrum observed at 280, 350 and 430 nm in three excited states. The calculated results are in excellent agreement with the experimental values. In addition, there is only one main absorption, as well as one emission wavelengths with the strongest oscillator strength. These wavelengths correspond to the π-π* excitation of the solely highest occupied (HOMO) to lowest unoccupied (LUMO) molecular orbital with the largest transitional proportion.
Homo and Lumo analysis
The Homo of the title molecule is localized on the unity of the azo and the Lumo is located on the oxime region as shown in Figure 5. However, Homo-2 and Lumo+1 are localized mainly on the benzene ring of the azo and oxime groups, respectively. Thus, the transition from Homo to Lumo is easier than that from Homo-1 to Lumo, Homo to Lumo+1, Homo-1 to Lumo+1 and Homo-2 to Lumo+2. This phenomenon also elucidates why the lowest energy absorption is a charge transfer transition from Homo to Lumo and mainly an electronic transition. Therefore, the electron density decreases significantly in the electron-donating azo when electrons transfer from the Homo to Lumo. This phenomenon is accompanied by an increase in the electron density of the electron-accepting the moiety. This result indicates that the electrons transfer from the unit of the azo to oxime group. The Homo and Lumo energies for PDBO calculated at -6.269502 eV and -2.509433 eV, respectively, where as the Homo-Lumo energy gap is 3.760069 eV.

Figure 5: The frontier molecular orbitals for PDBO.
In addition to this, the various quantum chemical parameters such as: Ionization potential, electron affinity, electrophilicity index, chemical potential, electro negativity and hardness are calculated using the standard procedure [28] and are listed in Table 6.
| Parameters | Values |
|---|---|
| HOMO | -6.269 eV |
| LUMO | -2.509 eV |
| Energy gap | -3.760 eV |
| Ionization potential (IP) | -6.269 eV |
| Electron affinity (EA) | -2.509 eV |
| Electrophilicity Index (ω) | 2.562 |
| Chemical Potential (µ) | 4.389 |
| Electro negativity (χ) | -4.389 eV |
| Hardness (η) | -3.760 |
Table 6: The Physico-Chemical properties of PDBO.
MEP analysis
The MEP surface map was calculated at B3LYP/6-311++G(d,p) method. MEP is related to the electronic density. It is very useful descriptor in understanding sites for electrophilic attacks and nuleophilic reactions as well as hydrogen bonding interactions [52]. The importance of MEP lies in the fact that it simultaneously displays molecular size, shape as well as positive, negative and neutral electrostatic potential regions in terms of colour grading as shown in Figure 6.

Figure 6: Molecular electrostatic potential map of PDBO.
Potential increases in the order red
Mulliken atomic charges
The mulliken atomic charges plot of the title molecule is shown in Figure 7. The charges was calculated at B3LYP/6-311++G(d,p) level of basis set and are listed in Table 7. It reveals that when compared to N12, N13 is considered as more basic site, which also support to the proton migration from hydroxyl group. The charge distribution shows that the more negative charge is concentrated on oxygen atom where as the partial positive charge resides at hydrogen. In PDBO the C19/C21 atoms have got most positive/negative charges, which are due to the attachment of oxime O23-H24 groups, respectively.
| Atoms | Charges | Atoms | Charges |
|---|---|---|---|
| 1C | -0.130 | 16C | -0.405203 |
| 2C | -0.068 | 17C | 0.051539 |
| 3C | -0.391 | 18H | 0.177496 |
| 4C | 0.155 | 19C | 1.752138 |
| 5C | -0.182 | 20H | 0.29974 |
| 6C | -0.358 | 21C | -2.218842 |
| 7H | 0.186 | 22H | 0.154298 |
| 8H | 0.159 | 23O | -0.211696 |
| 9H | 0.185 | 24H | 0.265845 |
| 10H | 0.185 | 25C | 0.546891 |
| 11H | 0.148 | 26H | 0.163336 |
| 12N | 0.089 | 27N | -0.375776 |
| 13N | 0.096 | 28O | 0.128423 |
| 14C | -0.924 | 29H | 0.227259 |
| 15C | 0.293 |
Table 7: The Mulliken charges of PDBO.

Figure 7: The Mulliken atomic charges of PDBO.
Temperature dependence of thermodynamic properties
Figure 8 depicts the correlation of heat capacity at constant pressure (Cp), enthalpy change (H0-E0)/T), Gibb’s free energy (G0-E0)/T, entropy (S), and (E) thermal energy with temperature by B3LYP/6- 311++G(d,p) method.

Figure 8: Thermodynamics and different temperatures of PDBO.
It cal be observed from the Table 8, that thermodynamic functions are increasing with temperature ranging (100 to 1000°K). This may be due to the fact that the different energy values in accordance with equipartition theorem corresponding to the 3N degrees of freedom may increase with increases in temperature, which in turn increase the internal energy of the molecules and thus the other thermodynamic parameters. The correlation equations between heat capacity, entropy, enthalpy changes and temperatures were fitted by quadratic fomula. The corresponding fitting factors (R2) 0.99952, 0.99999 and 0.99946, respectively and their corresponding fitting equations are as follows:
| T (K) | S (J/mol.K) | Cp (J/mol.K) | ddH (kj/mol) |
|---|---|---|---|
| 100 | 357.4 | 109.3 | 7.2 |
| 200 | 455.4 | 182.8 | 21.8 |
| 300 | 544.1 | 259.5 | 43.9 |
| 400 | 628.7 | 330.6 | 73.5 |
| 500 | 709.1 | 389.9 | 109.6 |
| 600 | 784.6 | 437.3 | 151.1 |
| 700 | 855 | 475.1 | 196.8 |
| 800 | 920.5 | 505.6 | 245.9 |
| 900 | 981.5 | 530.7 | 297.7 |
| 1000 | 1038.5 | 551.6 | 351.9 |
Table 8: Thermodynamic properties at different temperatures of PDBO.
C0 p,m=4.45407+0.0186T+1.64807 × 10-5 T2 (R2=0.99952)
S0 m=1.08583+0.00453T-4.01775 × 10-5 T2 (R2=0.99999)
ΔH0 m=3.73263+0.01559T+1.38113 × 10-5 T2 (R2=0.99946).
The title molecule was synthesized and characterized by spectral analysis such as FT-IR, FT-Raman and UV-Visible studies. A complete vibrational analysis has been performed for the first time for PDBO molecule. The bond parameters were calculated and compared with the related XRD data. The bond angles between C-N and benzene rings are equal: C2-C3-N12=C15-C14-C13=115˚ and C4-C3-N12=C16-C14-N13=124˚ which show there is no steric repulsion in between the Azo rings of PDBO. The -C-N=N-C- vibration indicate that the two unequivalent C-N parts (C3-N2, C14-N13), which causes a change in dipole moment and hence the azo group has increased double bond character. The β0 value of PDBO is forty five times larger than magnitude of urea; hence the molecule has good NLO property. The νO23-H24 mode observed at higher frequencies due to the high energy transfer from LPO23 to π*C17-C21 anti-bonding orbital. UV-Visible absorption analysis the Experimental band at 350 nm is attributed mainly due to a HOMO → LUMO transition is predicted as π-π* transition. The physicochemical properties and mulliken atomic charges were calculated. The thermodynamic functions have been plotted at different temperatures of PDBO molecule.