Organic Chemistry: Current Research
Open Access
ISSN: 2161-0401
ISSN: 2161-0401
Research Article - (2018) Volume 7, Issue 2
Background
Carbohydrate recognition has been shown to be involved in various biological processes. E-selectin is expressed on the endothelial cells stimulated by cytokines, such as IL-1β or TNF-α at inflammatory sites and plays an important role in the transport of neutrophils to inflammatory sites. The tetrasaccharide SLex is distributed on the surface of neutrophils and has been shown to be a ligand recognized by E-selectin. SLex and its derivatives can block the interaction between SLex and E-selectin and will be useful as the anti-inflammatory and anti-tumor agents.
Results
Random fucosylation on a partially protected sialyl LacNAc trisaccharide, resulted in the corresponding glycosylated products as a mixture. After careful purification, debenzylation, deacetylation, alkaline hydrolysis and further purification on C-18 column, a tetrasaccharide library containing five sialyl lewis X related tetrsaccharides was obtained, and the results were confirmed by proton NMR study.
Conclusion
Random fucosylation was demonstrated as efficient method to generate sialyl related tetrasaccharide library.
Keywords: Random fucosylation, Sialyl related tetrasaccharide, Oligosaccharide library
Carbohydrate recognition has been shown to be involved in various biological processes [1]. E-selectin is expressed on the endothelial cells stimulated by cytokines, such as IL-1β or TNF-α at inflammatory sites and plays an important role in the transport of neutrophils to inflammatory sites [2]. The tetrasaccharide SLex is distributed on the surface of neutrophils and has been shown to be a ligand recognized by E-selectin [3]. As the interaction between SLex and E-selectin is essential for the initial stage of neutrophil infiltration of the inflammatory site, sialyl lewis X tetrasaccharide and its derivatives, should be useful as new anti-inflammatory and anti-tumor agents [4].
Much research on the synthesis of sialyl lewis X related oligosaccharides and mimics was reported in order to find more active compounds [5], however, all of the reported processes are laborintensive, time consuming, and expensive, and there is a dearth of efficient methods for the synthesis of sialyl lewis X tetrasaccharide and its derivatives [6].
Random glycosylation on unprotected glycosylation acceptors generates oligosaccharide libraries containing all possible oligosaccharides with certain configuration [7]. For example, random fucosylation on three unprotected GlcNAc related disaccharides generated 18 fucosylated trisaccharides with α linkage [8], random galactosylation on an unprotected GlcNAc acceptor generated a library containing six possible disaccharides with α and β linkages [9] random glycosylation of glucosamine and galactosamine related donors on unprotected monosaccharide acceptors gave the β-linked GalNAc and GlcNAc related disaccharide libraries with excellent ratio [10]. In continuation of our studies on random glycosylation, we have been interested in generating sialyl lewis X tetrasaccharide and its derivatives. Herein, we like to report the synthesis of sialyl lewis X related tetrasaccharide library by random fucosylation of a partially protected trisaccharide as shown in Figure 1.
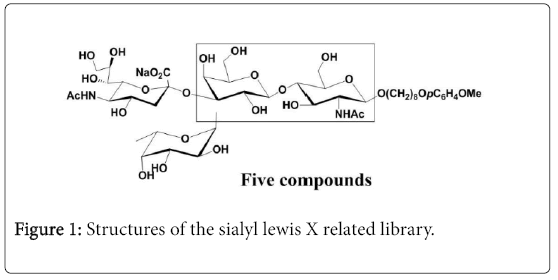
Figure 1: Structures of the sialyl lewis X related library.
TLC was performed on silica gel 60-F254 and charred with H2SO4. 1H NMR spectra were recorded at 300 MHz (Bruker AM-300), 360 MHz (Bruker AM-360), or 500 MHz (Varian UNITY 500) on solution in CDCl3 (internal TMS, δ0), or D2O (internal acetone, δ2.23), 13C NMR spectra were recorded at 75 or 90 MHz on the same instruments in CDCl3 (internal TMS, δ0) or D2O (internal dioxane, δ67.4). MALDI MS was obtained from Kratos Kompact MALDI. Methanol was distilled from Mg, and toluene was distilled from CaH2.
Procedures for preparation of compound 3
A solution of compound 2 (2.9 g, 2 mM) in dry THF (50 mL) was added TBAF (780 mg, 3 mM), and stirred at room temperature under nitrogen for 4 hours. After removal of the solvent, the residue was purified on a silica gel column eluted with hexane/ethyl acetate (1:1). The product was confirmed by LCMS and treated with CCl3CN in dry dichloromethane in the presence of dry K2CO3. After stirring under nitrogen at room temperature for 2 hours, the solid was filtered, and the solvent was evaporated under reduced pressure to give the product in more than 95% purity based on LCMS analysis, which was used to couple with HO(CH2)8OpC6H4OMe in dry dichloromethane in the presence of BF3•Et2O directly. After work up and purification, the desired disaccharide 3 was obtained in 40% over three steps as a white solid. 1H NMR (CDCl3, 300 MHz): δ6.75 (s, 4H), 5.30 (d, 1H, J=3.1 Hz), 5.15 (dd, 1H, J=10.34, 7.84 Hz), 4.90 (t, 1H, J=12.31 Hz), 4.89 (d, 1H, J=10.39 Hz), 4.42 (d, 1H, J=7.86 Hz), 4.41 (dd, 1H, 12.02, 1.69 Hz), 4.10-4.00 (m, 4H), 3.82 (t, 4H, J=6.40 Hz), 3.70 (s, 3H), 3.65 (t, 1H, J=9.50 Hz), 3.50 (m, 2H), 3.45 (dd, 1H, J=10.35, 9.04 Hz), 2.10 (s, 3H), 2.08 (s, 3H), 2.78 (s, 3H), 2.02 (s, 3H), 1.99 (s, 3H), 1.91 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ169.98, 169.98, 169.84, 169.71, 169.17, 168.67, 153.4, 115.2, 114.4, 101.6, 100.6, 77.4, 77.0, 76.6, 75.9, 72.5, 71.7, 70.8 70.5, 70.2, 68.9, 68.3, 66.5, 63.9, 61.8, 60.8, 55.4, 29,2, 29.1, 29.1, 28.9, 25.7, 25.5, 20.6, 20.5, 20.3, 20.2, 20.3, 20.3.
Procedures for preparation of compound 4
To the solution of compound 3 (0.9 g, 1.5 mM) in dry methanol was added catalytic amount of fresh prepared NaOMe, and the mixture was stirred at room temperature for 2 hours, few drops of acetic acid were added, and the solvent was removed under reduced pressure to provide the product which was confirmed by LCMS and NMR spectra. 1H NMR (D2O, 300 MHz): δ6.70 (s, 4H), 4.42 (d, 1H, J=7.80 Hz), 4.38 (d, 1H, J=7.86 Hz), 3.65 (s, 3H), 3.12 (t, 1H, J=7.81 Hz); 13C NMR (CD3OD/CDCl3, 75 MHz): δ153.4, 153.0, 115.3, 114.4, 103.5, 101.6, 79.9, 75.2, 74.5, 73.2, 73.1, 70.9, 70.1, 68.8, 68.6, 65.5, 61.2, 60.9, 55.4, 29.2, 28.9, 28.9, 28.9, 25.6, 25.6. The product was then reacted with 2,2- dimethoxypropane in the presence of 4Å molecular sieve and ptoluene sulfonic acid in acetonitrile for 2 hours to provide two isopropylidene protected derivatives, after work up and separation, the desired isopropylidene protected derivative was obtained and confirming by NMR spectrum and LCMS. 1H NMR (CDCl3, 300 MHz): δ6.80 (s, 4H), 4.59 (s, 1H), 4.40 (d, 1H, J=7.80 Hz), 4.31 (d, 1H, J=3.76 Hz), 4.39 (d, 1H, J=8.04 Hz), 3.85 (s, 3H), 1.50 (s, 3H), 1.18 (s, 3H); 13C NMR (CD3OD, 75 MHz): δ155.9, 154.0, 116.5, 115.6, 110.5, 104.2, 102.9, 80.8, 76.3, 75.3, 74.9, 74.7, 74.4, 70.9, 69.6, 67.7, 62.4, 61.7, 56.1, 30.7, 30.6, 30.4, 30.3, 28,4, 27.0, 26.5.70.1, 68.8, 68.6, 65.5, 61.2, 60.9, 55.4, 29.2, 28.9, 28.9, 28.9, 25.6, 25.6. The product was then treated with BnBr in dry DMF in the presence of NaH at room temperature for 4 hours, methanol was added to quench the reaction. After removal of the solvent under reduced pressure, the residue was purification on silica gel column with hexane and ethyl acetate to give the benzylated product, which was confirmed by LCMS and NMR studies. 1H NMR (CDCl3, 300 MHz): δ6.85 (s, 4H), 5.20 (d, 1H, J=10.55 Hz), 4.90 (d, 1H, J=11.15 Hz), 4.80 (d, 2H, J=10.90 Hz), 4.75 (s, 2H), 4.60 (d, 1H, J=12.11 Hz), 4.41 (d, 1H, J=7.80 Hz), 4.48 (d, 1H, J=12.10 Hz), 4.25 (d, 1H, J=7.90 Hz), 3.85 (s, 3H), 1.52 (s, 3H), 1.49 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ109.8, 102.2, 101.9, 81.3, 80.7, 79.4, 76.0, 75.2, 73.6, 73.4, 73.3, 73.2, 72.1, 70.1, 70.1,68.9, 68.6, 67.9, 65.9, 55.7. 31.6, 29.5, 29.3, 29.2, 28.0, 26.4, 25.9, 25.9. The product was treated with 80% acetic acid in water at 50°C for 3 hours, the solvent was removed under reduced pressure to give the desired diol 4 in 40% yield over four steps. 1H NMR (CDCl3, 300 MHz): δ5.10 (d, 1H, J=10.75 Hz), 4.89 (d, 1H, J=11.52 Hz), 4.81 (d, 2H, J=12.03 Hz), 4.68 (d, 1H, J=12.01 Hz), 4.65 (d, 1H, J=11.01 Hz), 4.51 (d, 1H, J=6.63 Hz), 4.50 (d, 1H, J=12.06 Hz), 4.43 (d, 1H, J=12.06 Hz), 4.30 (d, 1H, J=7.39 Hz), 4.10 (t, 1H, J=8.94 Hz), 3.80 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ102.4, 101.8, 80.8, 79.9, 76.0, 75.0, 74.8, 73.3, 73.0, 72.9, 71.9, 69.9, 68.6, 68.4, 67.8, 65.7, 55.5, 29.3, 29.1, 29.1, 29.1, 25.8, 25.7.
Procedures for preparation of compound 1
The mixture of the compound 4 and compound 5 in dry dichloromethane was stirred with 4Å molecular sieve for three hours, NIS (0.1 equiv.) and TfOH (0.1 equiv.) was added, and the mixture was stirred at -35C for four hours. Few drops of Et3N was added to quench the reaction, after evaporation, the residue was carefully purified on a silica gel column (eluted with CH2Cl2/MeOH) to provide the desired α-sialylated trisaccharide in 45% yield. 1H NMR (CDCl3, 300 MHz): δ7.50-7.10 (m, 20 H), 5.40 (dt, 1H), 5.35 (dd, 1H, J=7.90, 1.84 Hz), 5.21 (d, 1H, J=9.76 Hz), 4.84 (m, 1H), 4.78 (d, 1H, J=11.67 Hz), 4.65 (m, 2H), 4.58 (d, 1H, J=8.12 Hz), 4.53 (d, 1H, J=7.25 Hz), 4.51 (d, 1H, J=12.16 Hz), 4.45 (d, 1H, J=11.96 Hz), 4.37 (d, 1H, J=11.94 Hz), 4.34 (dd, 1H, J=11.95, 2.21 Hz), 4.20 (d, 1H, J=7.4 Hz), 4.1-3,9 (m, 5H), 3.90-3.80 (m, 4H), 3.81 (s, 3H), 3.79 (s, 3H), 3.77-3.66 (m, 2H), 3.55-3.47 (m, 4H), 3.40 (m, 3H), 2.79 (s, 1H), 2.50 (dd, 1H, J=12.92, 4.48 Hz), 2.39 (s, 3H), 2.08 (s, 3H), 2.01 (s, 3H), 2.00 (s, 3H), 1.90 (s, 3H), 1.87 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ102.2, 101.8, 81.2, 78.3, 76.2, 76.2, 75.9, 75.2, 74.9, 73.3, 72.9, 72.6, 69.9, 69.2, 68.9, 68.6, 68.6, 68.4, 67.9, 67.2, 65.8, 62.0, 55.6, 55.6, 53.0, 49.0, 31.0, 29.4, 29.2, 29.1, 29.1, 25.9, 25.7, 22.9, 21.3, 20.9, 20.7, 20.6. The product was then dissolved into the pyridine at room, H2S was bubbled into the reaction mixture for two days, the TLC showed the reduction reaction was finished. The solvent was evaporated under reduced pressure, and the residue was dissolved into methanol, acetic anhydride was added at room temperature, after stirred at room temperature for 2 hours, the mixture was removed and purified on a silica gel column to give the Nacetyl product. 1H NMR (CDCl3, 300 MHz): δ7.40-7.20 (m, 20 H), 6.75 (s, 4H), 5.75 (d, 1H, J=2.33 Hz), 5.39 (dt, 1H), 5.30 (dd, 1H, J=8.1 Hz), 5.15 (d, 1H, J=9.65 Hz), 4.89 (d, 1H, J=3,73 Hz), 4.88 (m, 1H), 4.85 (s, 1H), 4.74 (s, 2H), 4.60-4.40 (m, 8H), 4.30 (dd, 1H, J=12.45, 2.48 Hz), 4.10 (m, 13H, 3.81 (s, 3H), 3.80 (s, 3H), 3.75-3.40 (m, 14H), 2.80 (d, 1H, J=2.90 Hz), 2.50 (dd, 1H, J=13.02, 4.56 Hz), 2.09 (s, 3H), 2.01 (s, 3H), 1.98 (s, 3H), 1.92 (s, 3H), 1.89 (s, 3H), 1.83 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ102.6, 99.6, 98.4, 78.2, 77.5, 76.0, 74.9, 74.9, 74.9, 73.8, 73.3, 73.0, 72.7, 70.1, 69.3, 69.1, 68.9, 68.7, 68.6, 68.6, 68.0, 67.2, 62.3, 55.8, 55.8, 52.9, 49.1, 36.3, 29.6, 29.4, 29.2, 29.2, 25.9, 25.8, 23.4, 22.9, 21.3, 20.9, 20.7, 20.6. The product was then hydrogenated in methanol in the presence of Pd/C under hydrogen. After filtration through the celite, evaporation and purification on silica gel column, the partially protected sialyl LacNAc trisaccharide 1 was obtained in 28% yield over three steps. The product was confirmed by LCMS and NMR study. 1H NMR (CDCl3, 300 MHz): δ6.78 (s, 4H), 5.39 (m, 2H), 6.20 (s, 1H), 5.21 (dd, 1H, J=8.71, 1.47 Hz), 4.90 (dt, 1H, J=7.84, 1.47 Hz), 4.80 (s, 1H), 4.60 (d, 1H, J=8.19 Hz), 4.54 (d, 1H, J=7.79 Hz), 4.29 (dd, 1H, J=8.34, 2.26 Hz) , 4.05 (dd, 1H, J=10.39, J=12.31Hz), 4.00-4.87 (m, 4H), 4.83-4.78 (m, 3H), 3.75 (s, 3H), 3.70 (s, 3H), 3.65 (t, 1H, J=9.3 Hz), 3.58 (m, 1H), 2.65 (dd, 1H, J=12.96, 4.54 Hz), 2.10 (s, 3H), 2.08 (s, 3H), 2.00 (s, 3H), 2.00 (s, 3H), 1.99 (s, 3H), 1.89 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ103.8, 100.7, 97.8, 81.7, 74.7, 72.8, 72.4, 69.8, 68.7, 68.7, 68.7, 68.7, 68.7, 68.5, 68.4, 67.1, 62.6, 62.1, 62.6, 56.6, 55.8, 53.6, 49.6, 37.6, 29.7, 29.5, 29.4, 29.4, 26.0, 25.8, 23.5, 23.1, 21.3, 21.3, 20.8, 20.7. ESIMS: m/z 840 [M+H]+.
Procedures for random fucosylation on partially protected sialyl LacNAc trisaccharide
The mixture of the partially protected sialyl LacNAc trisaccharide (1) (420 mg, 0.5 mM) and per-O-benzyl fucosyl trichloroacetimidate (2) (570 mg, 1.0 mM) in dry DMF (5 mL) was stirred at room temperature under argon for 30 min. in the presence of CaSO4 (500 mg). After adding BF3•Et2O (30 mg), the mixture was stirred at room temperature for another 14 hours. Few drops of triethylamine were added to quench the reaction, and the reaction mixture was filtered through celite. The mixture was evaporated and purified on a silica gel column eluted with dichloromethane and methanol (10:1). Each fraction was checked by mass spectrometry, the unreacted acceptor and the donor hydrolysis products were removed. The combined tetrasaccharide mixture were hydrogenated in methanol in the presence of Pd/C for 48 hours at room temperature. After filtration through celite and evaporation, the residue was dissolved into the methanolic NaOMe. After stirring for 2 hours, few drops of water were added, and the mixture was stirred at room temperature for another 16 hours. The solvent was removed, and the residue was purified on C-18 column eluted with water and methanol to get the random glycosylation products. 1H NMR (D2O, 500 Hz) (Figure 2); ESIMS: m/z 826 [M+H]+.
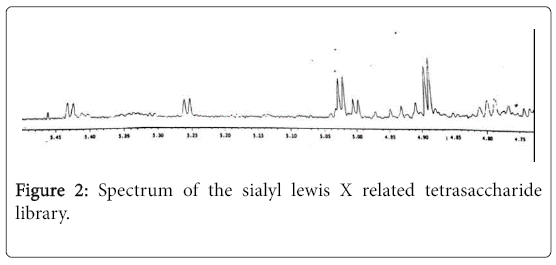
Figure 2: Spectrum of the sialyl lewis X related tetrasaccharide library.
The synthesis of library was initiated from the corresponding partially protected trisaccharide 1 which was synthesized from the commercially available compound 2 as was summarized in Scheme 1.
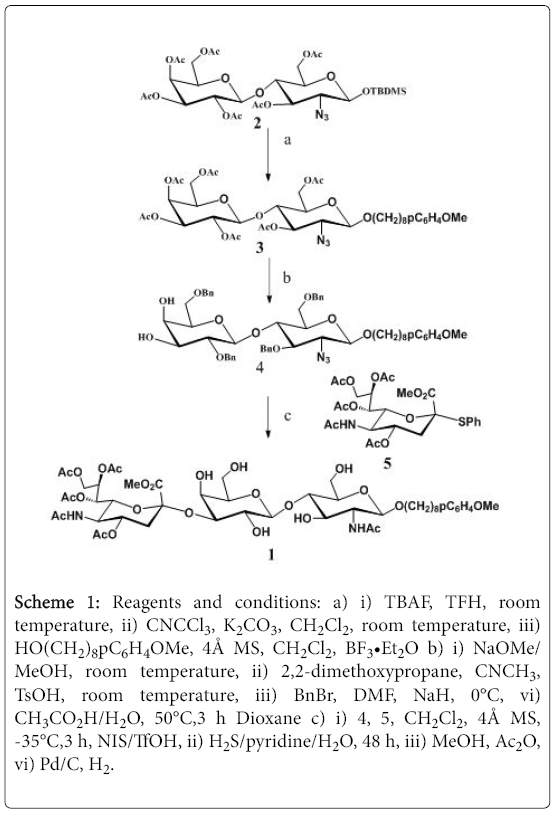
Scheme 1: Reagents and conditions: a) i) TBAF, TFH, room temperature, ii) CNCCl3, K2CO3, CH2Cl2, room temperature, iii) HO(CH2)8pC6H4OMe, 4Å MS, CH2Cl2, BF3•Et2O b) i) NaOMe/ MeOH, room temperature, ii) 2,2-dimethoxypropane, CNCH3, TsOH, room temperature, iii) BnBr, DMF, NaH, 0°C, vi) CH3CO2H/H2O, 50°C,3 h Dioxane c) i) 4, 5, CH2Cl2, 4Å MS, -35°C,3 h, NIS/TfOH, ii) H2S/pyridine/H2O, 48 h, iii) MeOH, Ac2O, vi) Pd/C, H2.
Thus, the compound 2 was treated by TBAF in THF for 2 hours, the desilylated product was then treated with trichloroacetonitrile in dry dichloromethane in presence of K2CO3 to provide the trichloroacetimidate, which was glycosylated with HO(CH2)8OpC6H4OMe to produce the disaccharide 3. The hydrophobic and UV active aglycon facilitates the purification of final library products by C-18 column or HPLC. After deacetylation with methanolic MeONa solution, isopropylidene formation from the reaction with 2,2-dimethoxypropane in acetonitrile in the presence of catalytic amount of TsOH, benzylation from the reaction with BnBr in THF in the presence of NaH, and removal of the isoproplylidene, the compound 4 was obtained, which was sialylated with the sialic acid donor 5, after reduction of the azido group with H2S/pyridine, Nacetylation with acetic anhydride in methanol, and debenzylation under hydrogen in the presence of Pd/C, the partially protected trisaccharide 1 was obtained, and after structural confirmation by MS and NMR study, the compound 1 was then used for the random fucosylation (Scheme 2).
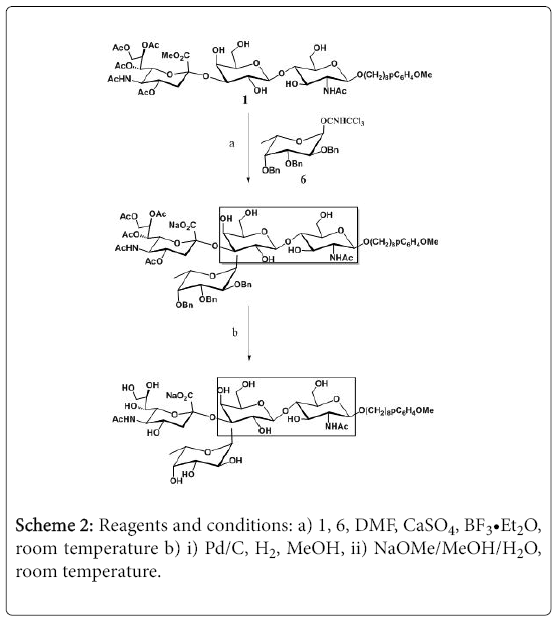
Scheme 2: Reagents and conditions: a) 1, 6, DMF, CaSO4, BF3•Et2O, room temperature b) i) Pd/C, H2, MeOH, ii) NaOMe/MeOH/H2O, room temperature.
Accordingly, the 2,3,4-tri-O-benzyl fucosyl imidate 5 was used as the glycosylation donor. To a mixture of compound 5 and compound 1 in dry DMF was added powdered dry CaSO4, and after stirring for 2 hours at room temperature under nitrogen, BF3•Et2O in dry dichloromethane was added, and the mixture was further stirred at room temperature for additional 10 hours. After quenching the reaction with few drops of triethyl amine, the solvent was removed under reduced pressure, and the residue was purified on a silica gel column (elution mixture: dichloromethane and methanol). The elution of each fraction was monitored by LCMS, and after taking into consideration of the recovered unreacted trisaccharide, the protected tetrasaccharide mixture was obtained in 35% yield (based on the conversion of the trisaccharide acceptor 1).
The tetrasaccharide mixture was debenzylated under hydrogen in presence of Pd/C (monitored by proton NMR spectroscopy for reaction completion). After the reaction was finished, the mixture was filtered through celite, evaporated, and purified on a silica gel column. The products were then dissolved in methanol and treated with freshly prepared NaOMe. After stirring at room temperature for 2 hours, few drops of water were added, and the solution was stirred at room temperature for additional 16 hours. The reaction was neutralized by addition of few drops of acetic acid. The volatiles were removed under reduced pressure, and the residue was purified by C-18 column to obtain the target tetrasaccharide library.
The tetrasaccharide library was confirmed by proton NMR and mass spectra. The proton NMR spectrum (Figure 2) shows five peaks for five α-linked fucosylated tetrasaccharides with a reasonable ratio. However, separation of these tetrasaccharides either by C-18 column or HPLC was found to be difficult.
In conclusion, random fucosylation on a partially protected sialyl trisaccharide, after deprotection and purification, a library containing all possible five sialyl lewis X related tetrasaccharides was efficiently obtained. The library was confirmed by mass and NMR spectral studies. The current study demonstrated that the random glycosylation is the most efficient way for the synthesis of sialyl lewis X related oligosaccharide libraries, and further work on synthesis of more complicated sialyl lewis X related oligosaccharide library is in progress.