Organic Chemistry: Current Research
Open Access
ISSN: 2161-0401
ISSN: 2161-0401
Mini Review - (2022)Volume 11, Issue 1
Palbociclib, Ribociclib and Abemaciclib are the drugs categorized as CDK4 and CDK6 cyclin-dependent kinase inhibitors. The current article reviews the patent and non-patent journals regarding the synthetic routes and final polymorphic forms of these CDK4/6 inhibitors. Palbociclib (PD0332991, Ibrance®), Ribociclib (LEE011, Kisqali®) and Abemaciclib (LY2835219, Bemaciclib, Verzenio®) are the drugs approved as cyclin-dependent kinase inhibitors (CDK4/6) and are used for the treatment of certain kind of breast cancer. Other reported CDK4/6 inhibitor is Trilaciclib (G1T28) which is still in phase 2 clinical trials. The drug Ribociclib was approved by USFDA on March 13, 2017, Palbociclib was approved by USFDA on March 31, 2017 and Abemaciclib was approved by USFDA on sept.28, 2017.
Palbociclib; Ribociclib; Abemaciclib; CDK4/6 inhibitors; Synthetic review; Cyclin-dependent kinase inhibitors; Breast cancer.
Uncontrolled proliferation of cells is trademark of cancer and thorough understanding of the cell division will help to regulate the cancer. There are different phases in a cell cycle starting from resting phase (G0) to interphase (G1, S, G2) and then to cell division (M). The enzyme CDK’s (4 and 6) controls the transition between the G1 and S phases of the cell cycle. During S-phase of cell cycle new DNA is synthesized which prepare itself to divide during the process of mitosis (M). Also, a tumor suppressor protein (as retinoblastoma protein, Rb) is dysfunctional in several major cancers and it also prevents the excessive cell growth. When cell division is about to start Rb is phosphorylated and it thus becomes inactive. These CDK4/6 inhibitors dephosphorylate the Rb protein, resulting in cell-cycle arrest and thus inhibit the proliferation of cancerous cells. Out of these three drugs, Palbociclib an oral agent by Pfizer categorized as BCS-II drug is used in combination with letrozole as hormone therapy for the first line treatment of post-menopausal women with ER+/HER2 metastatic breast cancer (Figure 1). It is referred as “breakthrough therapy” based on its good clinical performance in phase-III and thus resulted in fast track approval by USFDA in Feb.2015. Palbociclib is available as 75 mg, 100 mg, and 125 mg capsules. Combination therapy of Palbociclib with doxorubicin has shown the cumulative cytotoxic effects in RBproficient triple-negative breast cancer cells. Abemaciclib by Eli Lilly, categorized as BCS class-II drug received fast track approval from USFDA for the treatment of post-menopausal women with HR+/ HER2 advanced or metastatic breast cancer based on its clinical trial data [1]. For the treatment of post-menopausal women with HR+/ HER2 advanced or metastatic breast cancer abemaciclib is approved in combination with Aromatase Inhibitor (AI) whereas when combined with fulvestrant it was approved for the treatment of women with HR+/HER2 advanced or metastatic breast cancer. Based on the clinical trial data, abemaciclib-letrozole/anastrozole combination significantly reduced the risk of death or progression as compared to its combination with Non-Steroidal Aromatase Inhibitors (NSAI). The trail also indicated diarrhoea, neutropenia, and fatigue as most common adverse event. Abemaciclib is available in four tablet strengths as 50 mg, 100 mg, 150 mg and 200 mg. Another orally active agent categorized as BCS-IV drug by Novartis is Ribociclib which received USFDA approval on March 13, 2017 as initial endocrine-based therapy for the treatment of post-menopausal women with hormone receptor HR+/HER2 advanced or metastatic breast cancer [2]. Approval was based on randomized double blind, placebo controlled international clinical trial data. Ribociclib was also approved in Europe as a first line treatment for HR+/HER2 locally advanced or metastatic breast cancer in combination with any aromatase inhibitor. It is available as 200 mg film coated tablet and as 200 mg tablet in combination with letrozole (2.5 mg) known as Kisqali-Femara® co-pack.

Figure 1: The structure of inhibitor drugs.
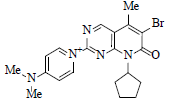
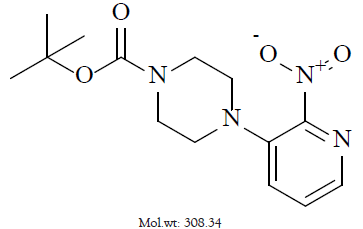
Palbociclib, a non-hygroscopic yellow to orange crystalline powder is marketed under the trade name Ibrance® was discovered by Pfizer and approved on March 31, 2017. Chemically it is free base and designated as 6-acetyl-8-cyclopentyl-5-methyl-2-{(5-piperazin-1-ylpyridin-2-yl) amino} pyrido(2,3-d) pyrimidin-7-one (Figure 2).

Figure 2: Palbociclib.
Drug discovery route to palbociclib
The drug discovery route reports the synthesis of hydrochloride salt of Palbociclib. This route involved the coupling between compound 2 and 3 to afford the intermediate 4, which followed by Stille coupling for C-C bond formation using the vinyl tin reagent lead to intermediate 5.
Deprotection strategy of 5 using HCl (g) in dichloromethane resulted in Palbociclib Hydrochloride 6. In-spite of highly convergent drug discovery route with 27.3% overall yield, it has few limitations such as coupling of 2 and 3 was highly precarious with only 38% yield, second the use of highly expensive and toxic tin reagent for the Stille coupling. The reaction pathway is shown in scheme 1.

Scheme 1: Drug discovery route to palbociclib.
First process chemistry route for manufacturing palbociclib
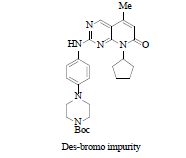
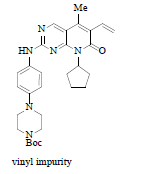
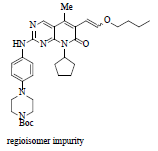
Drug discovery route laid the foundation of further process development activities and by using this route material for clinical trials was generated. Key highlights for this route involved: a) use of 7 instead of 2 for the process development activities, b) for SNAr coupling between compound 3 and 7 use of strong base such as LiHMDS (Lithium Hexamethyldisilazide), which significantly improved the yield, c) for C-C bond formation replacement of Stille coupling with Heck coupling, this help to eliminate highly expensive and toxic vinyl tin reagent, d) use of isethionic acid for the salt formation and thus isolating the free base of compound 6 as isethionate salt 9, this isolation facilitated the hydrolysis of enol ether and easy deprotection of tert.-butoxy carbonyl group. Nevertheless, the process improvement results were satisfactory but still few concerns need to be addressed before commercialization such as, i) quantity of compound 3, more than two equivalents are required for good yield, ii) challenge to control regioselective impurity, des-bromo impurity and vinyl impurity during Heck reaction (observed impurity level during lab study around 1% of des-bromo impurity, and 0.2%-0.4% of vinyl impurity, 1.2% regioisomer was observed when no additives were used in the reaction but with additives such as LiOTf (Lithium Trifluoromethane Sulfonate) level of regioisomeric impurity reduced to 0.3%-0.6%,) (moreover, to eliminate this impurity above level of 1.0% Simulated Moving Bed (SMB) chromatographic technique was used to achieve the specification of API as per ICH guidelines) iii) the most important was physico-chemical properties of isethionate salt 9. In spite of having very high aqueous solubility and other advantageous solid-state properties such as melting point, crystallinity and low hygroscopicity, still it was not compatible for formulation thus, the approach was again changed to free base for commercialization. The entire reaction pathway is depicted in Scheme 2.

Scheme 2: First process chemistry route for palbociclib isethionate salt.
List of impurities during heck reaction (stage-02)
Although change of Stille coupling to Heck coupling was a considerable achievement, even though lot of impurities were formed during the process development activities as listed in Table 1:
| Structure of Impurity | Observed limit during lab studies |
|---|---|
|
About 1.0% is formed when the product was exposed to i-PrMgCl via metal-halogen exchange reaction. |
|
About 0.2%-0.4% |
|
About 0.1%-0.3% |
Table 1: List of impurities during heck reaction.
To control these impurities to a substantial level during the process development studies, various routes were evaluated but none of these routes were cost effective and suitable for commercial manufacturing. Therefore, it was decided to go with same possibility with strong focus on safety during scale-up as well as commercial viability, scalability and quality of final product which should comply with ICH guidelines [3- 5].
Second process chemistry route for manufacturing palbociclib
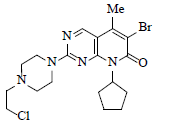
Thorough analysis of the first process chemistry route and other schemes it was decided to work on the same route (scheme-2) with possible modification to enable it for commercial manufacturing. This strategy mainly focused on the coupling reaction between 3 and 7. The SNAr (nucleophilic aromatic substitution reaction seems to be very easy in approach, but relatively complicated mechanism is involved in it. Comprehensive study was carried out to identify the appropriate conditions for coupling which should not only comply with cost it should also have the commercial viability. Lot of studies were carried out just to select the leaving group in 7 at 2nd position, such as methane sulfinyl, chloro, but by careful analysis of all the process research studies it was evident that chloro at 2nd position was highly effective as compared to methane sulfinyl group. Also, the synthesis of compound 7 can easily be achieved from commercially available dichlorobromopyrimidine (10) in four easy steps (scheme 3).

Scheme 3:Synthesis of compound 7.
Case study on stage-01
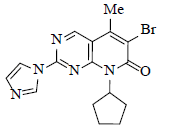
Alternative approach to coupling between 3 and 7 was the parameter to study. Various conditions were evaluated such as palladium catalysed (using palladium catalyst such as Pd(OAc)2, Pd2(dba)3, POPd along with ligands like BINAP, Davephos, Johnphos) SNAr coupling under neutral conditions, acidic conditions, weak basic conditions (using Pyridine, Hunig’s base, N-methylmorpholine, DMAP, Imidazole, DABCO, K2CO3, Cs2CO3), strong basic conditions (using LiHMDS) fluoride catalysed (fluoride catalyst such as CsF, KF in the presence of crown ethers) coupling.
Based on the mechanistic understanding of the coupling step and all the process development studies, Grignard base (under the category of strong base) was opted. This Grignard base by using two equivalents gave the comparable yield as was achieved by using LiHMDS with cleaner reaction profile and no dimer impurity. By stoichiometric conditions yield of coupling reaction increased to 94%. The selected Grignard reagent was i-PrMgCl (isopropyl magnesium chloride). Since it generates propane gas during reaction which may affect the scalability of the project, thus other Grignard reagents were studied with conclusion of using Cylcohexylmagnesium Chloride (CyMgCl) in THF solvent. This reduced the costing and commercialization constraint. Reaction pathway in scheme 4 summarises the process development activities for second process chemistry route.

Scheme 4:Stage-01 optimised snar mediated coupling reaction.
List of impurities during snar coupling reaction (stage-01)
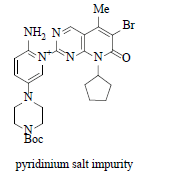
Since lot of process development activities was performed to achieve a scalable commercial process, thus the impurities formed during these activities are listed in Table 2:
| Structure of Impurity | Condition of formation |
|---|---|
|
Under neutral conditions (since the 3 is ambident nucleophile, suggesting the pyridine nitrogen as nucleophile), also under acidic condition, under weak basic condition (such as Pyridine, Hunig’s base, N-methylmorpholine, K2CO3, Cs2CO3 |
|
Under weak basic condition using 4-DMAP |
|
Under weak basic condition using imidazole |
|
Under weak basic condition using DABCO |
|
During fluoride catalysed conditions when CsF or KF is used along with 18-crown-6 or 15-crown-5 |
|
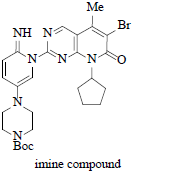
Imine Compound is also formed under strong basic condition when LiHMDS was used |
|
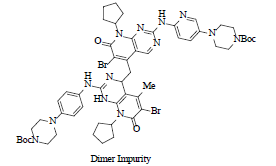
Dimer impurity formed during the use of LiHMDS base |
|
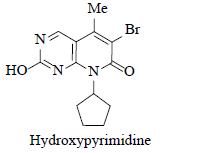
Reaction of 7 with water when strong bases such as NaOt-Bu, KOt-Bu and NaOt-Am were used. |
|
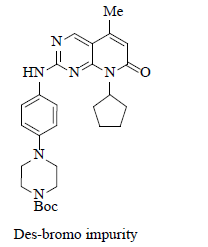
Des-bromo impurity is formed when the product was exposed to i-PrMgCl via metal-halogen exchange reaction. |
Table 2: List of impurities formed during coupling reaction.
Case study on (stage-02) heck reaction
To have control on all the key impurities observed during the lab studies of Heck reaction (first Process chemistry route), numerous studies were performed such as crystallization of compound 5 or crystallization of API in different solvents followed by investigational studies for different types of catalyst for Heck reaction [6]. The reactions were experimented by loading 5% of Nickel catalyst with two different bases (TEA and K2CO3) in 1-butanol at 90°C, but none of the conditions were found to be effective resulting only unreacted starting material as isolated product. Around 18 different types of Pd-C without ligand were tried in bases like TEA, DIPEA and DCHA at 90°C in 1-butanol as solvent media. The result showed only the starting material and desbromo impurity around 5%-33%. Different types of ligands, with 4% of Palladium loading in n-butanol at 90°C were performed and out of 250 experiments about 65 completed with yield more than 90%. The ligands such as Xantphos, bis (2-diphenylphosphinophenyl) ether (DPEPphos), ( ± ) BINAP, 1,1’-bis(di-i-propylphosphino)ferrocene (dippf), and 1,4-bis (diphenylphosphino) butane (dppb) were found to be effective in these completed reactions. Further optimization studies with varied parameters such as quantity of ligand, nature and quantity of base, solvent and its’ volume, reaction time and temperature, method of quenching, isolation procedure, loading of ligand, mole equivalent of n-butyl vinyl ether, quantity and loading of palladium catalyst, yield, presence or absence of moisture, impact of nitrogen, content of impurities in the isolated product, recovery of palladium and thereof lead to conclusion of reaction pathway as per scheme 5.

Scheme 5: Stage-02 optimised heck coupling reaction.
Case study on stage-03 (deprotection)
This study was based on deprotection of compound 8 and polymorphism/particle size of Palbociclib 15 (free base of compound 6). The aim of the study was to develop API (Palbociclib) of appropriate quality standards which can ease the formulation process and achieve good results in its bio-studies. During initial developmental studies, conversion of compound 8 to final API (Palbociclib) was easily achieved at low pH levels in aqueous solvents system followed by pH adjustment, resulted in small particle size (around 1-5 μm). This particle size reflected high degree of stickiness during formulation studies. To overcome this issue, recrystallization study was carried out by using variety of solvents such as n-butyl acetate, n-butanol, toluene, trifluorotoluene, chlorobenzene, DMF, NMP, anisole, pyridine, sulfolane and thereof. Potential stability issues observed when crystallization studies were performed in NMP and NMP/nbutanol mixture. After careful observation of crystallization studies, n-butanol/anisole system was selected based on the particle size and nature of compound so obtained which facilitated the drug product development [7]. Also, anisole belongs to class III solvent so limit was not a constraint to achieve as per ICH. With this crystallization system, Palbociclib of around 12-14 μm particle size was achieved. Scheme 6 depicts the stage-03 conclusive scheme.

Scheme 6: Stage-03 optimised deprotection strategy.
Third process chemistry route for manufacturing Palbociclib (WO2016030439A1 (EP 3186252)
As per the process reported, it shows the modification in intermediate synthesis, use of LiHMDS in toluene for coupling in stage-01, use of n-butyl vinyl ether for Heck reaction in n-butanol for stage-02 and hydrochloric acid in ethanol followed by sodium hydroxide for the isolation of free base, Palbociclib. It mainly emphasises on the large particle size of Palbociclib using n-butanol/anisole mixture at 95°C. Various impurities were reported during the synthesis of compound 18, such as Impurity A-1 and Impurity B-1. These impurities are listed in Table 3:
| Impurity A-1 | Impurity B-1 |
|---|---|
|
|
Table 3: Impurities which were reported during the synthesis of compound 18.
Synthesis of compound 12 requires the seeding material. The entire reaction pathway is shown in Scheme 7 and 8:

Scheme 7: Final scheme for second process chemistry route.

Scheme 8: Third process chemistry route for manufacturing large size palbociclib.
Fourth process chemistry route (US20170247380)
A generalized reaction scheme is shown below Scheme 9. This scheme indicates the ring closure reaction of 20 (wherein X represents all halogens/methane sulfinyl/methane sulfonyl) with 21 (wherein R represents methyl/ethyl/propyl/isopropyl/allyl/benzyl) to afford 22, which is recrystallized with hexane/ethyl acetate (1:1). The yield of the process where X represents chloro is around 91.6% whereas yield is 84.9% when X represents methyl sulfinyl group. For the synthesis of compound 24 (through substitution reaction) lot of solvents were tried such as dichloromethane (Preferred), Chloroform, EDC, acetonitrile, methyl benzene, THF, dioxane, DMC and the acid-binding agents such as caesium carbonate, potassium carbonate, lithium carbonate, sodium and potassium tert-butoxide, sodium Hydride (Preferred). For the conversion of 24 to 15 solvents studied were methylbenzene and xylene (preferred), DMF, DMAc, DMSO whereas for acidic hydrolysis, hydrochloric acid (Preferred), vitriol, phosphoric acid, acetic acid, TFAA and thereof were studied [8-10].

Scheme 9: General scheme for the synthesis of palbociclib.
This scheme has few limitations with respect to commercialization such as conversion of 20 to 22 explains the use of microwave oscillator reaction system. This is commercially not viable. Use of sodium hydride, lithium (bistrimethylsilyl) amide at commercial level is cumbersome and difficult to handle. Also, the process has not disclosed any HPLC purity of the compound.
Fifth process chemistry route (US2017247379A1)
The reported process is represented as in Scheme 10. This explains the use of commercially available raw materials such as malononitrile 25, 2-acetyl-2-butenoic acid methyl ester 26, halogenated cyclopentane intermediate as 27, N-(5-(1-piperazinyl)-2-pyridinyl) guanidine 30, reagents such as sodium selenite for dehydrogenation reaction. The overall yield of the process is 32.8% but it has not disclosed any information related to HPLC Purity. The compound 26 is subjected to ring closure metathesis with malononitrile followed by substitution reaction with 27 to afford 29. This is reacted with N-(5-(1-piperazinyl)-2- pyridinyl) guanidine 30 to get 31 which upon dehydrogenation at high temperature 150°C-160°C for 5-6 hrs using sodium selenate affords Palbociclib 15.

Scheme 10: Ring closure metathesis for the synthesis of palbociclib.
Sixth process chemistry route (WO2016016769A1)
In this process silyl derivative of crotonic acid (32) was used as a novel intermediate. This derivative 32 was reacted with 12 in the presence of suitable palladium catalyst and base (with ligand as an option) to afford 33 which upon intramolecular cyclisation resulted in compound 13 [2]. Conversion of 13 to Palbociclib 15 follows the prior art process. The entire reaction pathway is shown in Scheme 11.

Scheme 11: Silyl derivative of crotonic acid as novel intermediate for palbociclib.
Seventh process chemistry route (WO2018/007927A1)
The reported one pot process for the synthesis of 15 comprises reaction between 7 and 3 in THF at -10°C using 2 M THF solution of isopropyl magnesium chloride, Pdcl2(dppf), DIPEA and n-butyl vinyl ether. Compound 8 so obtained is converted into methane sulfonic acid salt 33 of form A and form B in methanol-water at 55-60°C. This process also reports various acid salts of Palbociclib such as oxalate salt, trifluoroacetate salt, isethionic acid salt and thereof. The overall strategy of the process is novel acid salts of Palbociclib (characterized by XRD values) and basification of these salts to afford large particle size (surface area more than 2 m2/g) to ease the filtration/isolation and drug product development process. Reported purity of Palbociclib is more than 99%. The reaction pathway is shown in Scheme 12.

Scheme 12: Novel methane sulfonate salt of compound 15.
Salts and polymorphism in palbociclib (wo2005005426, wo2016066420a1, wo2016127963a1, wo2016024249a1, wo2014128588, us7863278)
Palbociclib is stable as free base but different salts are also reported such as isethionate, methane sulfonate, oxalate, di and monohydrochloride, trifluoroacetate and thereof. Palbociclib in the form of free base exhibits polymorphism and exists in form A and B. Form A is anhydrous crystalline and thermodynamically more stable than form B, thus it was opted for developmental studies and commercialization. Form A is characterized by 2θ values at 5.1, 8.0, 10.1, 10.3, 11.5, 14.0, 15.1, 16.0, 17.1, 18.7, 19.7, 20.2, 21.2, 22.5, 23.0 ± 0.2 whereas form B is characterized by 2θ values at 6.0, 10.9, 12.8, 16.4, 19.8, 18.1, 22.6, 26.7, 28.2 ± 0.2.
Mono-isethionate salt of Palbociclib exists in more than one polymorphs such as Form A, Form B and Form D. Each form has its own characteristic XRD pattern, DSC and Raman spectra value in Table 4:
| Polymorphic Forms | Characteristic XRD (2θ values) | DSC (°C) | Raman (cm-1) |
|---|---|---|---|
| Form A | 8.7, 13.5, 17.6 | sharp endotherm at 273 | 1600, 1290, 675, 470, 450, 425 |
| Form B | 5.1, 10.2, 11.8, 12.1, 12.8, 13.1, 14.7, 15.2, | sharp endotherm at 271 | 1600, 1290, 470, 450, 425 (absence of substantial peak at 675) |
| Form D | 8.4, 8.9, 21.9 | sharp endotherm at 277 | 463 |
Table 4: Each form has its own characteristic XRD pattern, DSC and Raman spectra value, are shown.
Palbociclib dihydrochloride exhibited adequate water solubility but in the low humidity conditions (around 10%), its moisture uptake was high, approximately greater than 2% of its mass thus resulting this form unsuitable for formulation use. Palbociclib monohydrochloride was reported as marginally hygroscopic with moisture uptake of greater than 2% of its mass in heavy humidity condition (around 80%). The monohydrochloride salt existed in Form-1, Form-2, Form-3, Form-4, Form-5, Form-6, Form-7 and amorphous form. Form-1 is characterized by appropriate 2θ values (in XRD diffractogram) at 4.7, 9.9, 11.19, 15.6, 15.8, 19.2, 22.7 and 24.3 ± 0.2. Form-2 is anhydrous, relatively less hygroscopic, chloride content of 7.39% and is characterized by 2θ values at 9.9, 11.5, 14.9, 16.3, 19.0, 21.7, and 22.2 ± 0.2. Form-3 is non-hygroscopic, anhydrous form with water uptake of about 0.70% between 20% and 70% RH and is characterized by 2θ values at 9.4, 11.8, 16.3, 18.8, 22.4, 22.8 and 23.7 ± 0.2. Form-4 is obtained by evaporation process using mixed solvent system (acetonitrile: water, 9:1) and is characterized by 2θ values at 3.35, 4.78, 7.80, 12.16, 18.64, 20.16 and 22.18 ± 0.2. Similarly Form-5 is isolated by crash-cooling from THF: Water mixture at 2°C and is characterized by 2θ values at 4.47, 6.45, 17.95, 18.14, 19.46, 22.23 and 22.71 ± 0.2. Form-6 is isolated by evaporation process using mixed solvent system THF: Water (85:15) and is characterized by 2θ values at 2.79, 4.73, 7.62, 8.17, 11.81, 19.63, 20.01 and 21.60 ± 0.2. Form-7 is characterized by 2θ values at 4.34, 9.88, 10.02, 15.63, 19.85, 21.00 and 23.69 ± 0.2. Form- 2 was extensively used for the formulation study purpose.
The other reported salts when Palbociclib and respective acid (1:1) are tabulated below (Table 5):
| Salt | XRD (2θ values) | DSC (°C) |
|---|---|---|
| Palbociclib Hydrobromide | 11.5, 14.8, 16.5, 19.1 and 21.9±0.2 | 315 |
| Palbociclib sulphate | 4.1, 9.3, 11.7, 16.2, 17.2 and 21.2±0.2 | 273 |
| Palbociclib oxalate | 4.5, 10.0, 13.8, 17.3, 19.6 and 23.1±0.2 | 256 |
| Palbociclib besylate | 9.0, 10.7, 11.9, 15.8, 22.1 and 26.2±0.2 | 294 |
| Palbociclib salicylate | 6.9, 11.7, 13.9, 16.9, 21.8 and 26.2±0.2 | 258 |
| Palbociclib fumarate | 5.1, 7.6, 12.9, 17.8 and 24.9±0.2 | 197 |
| Palbociclib 2,4-dihydroxybenzoate | 6.3, 10.6, 12.7, 16.4, 19.2 and 21.3±0.2 | 214 |
| Palbociclib benzoate | 4.1, 8.8, 14.0, 18.9, 21.6 and 23.8±0.2 | 221 |
| Palbociclib mesylate | 9.9, 13.4, 19.4 and 21.6±0.2 | 312 |
Table 5: The other reported salts when palbociclib and respective acid is in 1:1 are shown.
The other reported salts when Palbociclib and respective acid is in 1:2 are shown in Table 6.
| Salt | XRD (2θ values) | DSC (°C) |
|---|---|---|
| Palbociclib Hydrobromide | 9.4, 11.5, 16.3, 19.2, 20.4 and 22.0±0.2 | 207 |
| Palbociclib sulphate | 3.1, 9.1, 12.6, 16.0, 20.1 and 26.6±0.2 | 283 |
| Palbociclib besylate | 6.3, 8.2, 11.4, 14.3, 17.8 and 20.6±0.2 | 283 |
| Palbociclib mesylate | 6.9, 9.7, 13.9, 16.6, and 19.6±0.2 | 302 |
Table 6: The other reported salts when palbociclib and respective acid is in 1:2 are shown.
Palbociclib also exists in various crystalline forms as summarised below in Table 7:
| Form | XRD (2θ values) | DSC (°C) |
|---|---|---|
| I | 4.3, 6.3, 7.9, 8.5, 12.3, 16.9, 20.3, 22.9, 24.7, 27.4, 31.7±0.2 | Endothermic peaks at about 81.3, 155.2, 238.0, 265.3 and exothermic peak at about 185.7. |
| II | 5.1, 8.0, 10.1, 10.3, 11.5, 14.0, 15.1, 15.9, 16.5, 17.0, 17.4, 17.9, 18.7, 19.6, 20.2, 20.5, 21.1, 21.9, 22.4, 22.9, 23.7, 26.2, 27.4, 28.4, 28.8, 29.4, 30.0, 30.7, 31.7, 32.9, 34.6, 35.8, 39.2±0.2 | Endothermic peaks at about 78.0 and exothermic peak at about 275.1. |
| III | 5.1, 7.9, 8.5, 10.1, 10.2, 11.5, 14.1, 15.2, 15.8, 15.9, 16.4, 17.0, 17.4, 18.7, 19.6, 20.2, 21.0, 22.2, 22.4, 23.7, 25.9, 27.2, 28.4, 30.0, 30.7, 31.0, 32.8, 34.6±0.2. | Not reported |
| IV | 5.1, 8.0, 10.2, 11.5, 14.1, 15.2, 16.0, 17.1, 17.7, 18.6, 19.7, 20.4, 22.3, 23.3, 24.1, 26.0, 28.2, 30.1, 30.6, 34.6, 35.9, 37.5, 38.3, 39.3±0.2 | Not reported |
| V | 5.7, 7.8, 8.9, 11.5, 13.1, 16.6, 18.3, 20.1, 21.2, 24.0, 26.7, 31.6±0.2 | Endothermic peaks at about 62.6, 126.0, 262.8. |
| V-A | 5.7, 7.8, 8.3, 8.8, 11.4, 12.4, 13.1, 14.7, 16.1, 16.6, 17.1, 17.6, 18.4, 19.5, 20.0, 20.6, 21.1, 21.4, 22.2, 24.0, 24.9, 26.7, 27.3, 27.8, 29.2, 30.3, 31.5, 32.3, 34.5, 35.4, 37.3, 38.8, 39.6±0.2 | Endothermic peaks at about 148.2, 269.2, 272.1, 284.2 and exothermic peak at about 222.9. |
| VI | 5.7, 7.9, 9.0, 10.1, 10.5, 11.6, 13.4, 16.3, 17.0, 18.2, 20.6, 22.1, 23.9, 24.7, 25.3, 25.9, 27.7, 29.9, 32.2, 34.3, 35.5, 39.0±0.2 | Not reported |
| VII | 4.5, 8.0, 8.9, 10.2, 13.4, 15.6, 16.1, 17.2, 17.8, 19.1, 20.4, 20.8, 21.3, 22.3, 23.6, 24.1, 25.0, 25.7, 27.2, 28.3, 29.5, 31.4, 33.2, 35.2, 36.1±0.2 | Endothermic peaks at about 98.5, 251.2, 269.3 and 285.0. |
| VIII | 5.1, 7.9, 8.5, 9.5, 10.2, 11.4, 13.1, 14.0, 15.1, 15.7, 16.4, 16.9, 18.6, 19.0, 19.6, 20.3, 21.0, 22.2, 22.9, 23.7, 25.8, 27.2, 28.2, 29.8, 30.7, 31.7, 32.7, 34.7±0.2 | Not reported |
Table 7: Palbociclib also exists in various crystalline forms as summarised.
Palbociclib summary
Various synthetic procedures are reported in the literature, but it would be unfair to justify the best possible methodology for commercial manufacturing. So far, second process chemistry route using cylcohexylmagnesium chloride followed by Heck coupling with n-butyl vinyl ether is considered best for commercialization as it has been thoroughly studied for every step and parameter. This route also avoids the hazardous and toxic reagents. The process was able to achieve the best stable polymorph with large particle size to ease the formulation development. Various other studies regarding the polymorph were done and have been compiled. A through scientific understanding with critical analysis is required to have best possible route for commercial manufacturing with large particle size and stable polymorphs [11].
Ribociclib 36, an orally active agent developed by Novartis and Astex pharmaceuticals is a yellow to brown crystalline powder, slightly hygroscopic in nature and soluble in aqueous acids. It is available as succinate salt of free base in ratio 1:1 chemically designated as butanedioic acid-7-cylcopentyl-N, N-dimethyl-2-{(5-(piperazin- 1-yl) amino}-7H-pyrrol(2,3-d) pyrimidine-6-carboxamide having polymorphic form A, structure is shown below Figure 3.

Figure 3: Ribociclib succinate.
Drug discovery route
The disclosed drug discovery route follows the “Buchwald method B” wherein the compound 2-chloro-7-cyclopentyl-7H-pyrrolo(2,3-d) pyrimidine-6-carboxylic acid dimethylamide, 34 (300 mg) is reacted with 5-piperazin-1-yl-pyridin-2-ylamine, 35 (314 mg) to afford Ribociclib 36 as free base in 36% yield (142 mg). The entire chemical transformation is shown in Scheme 13:

Scheme 13:Drug discovery route for ribociclib.
To the mixture of 34 and 35, added Pd2(dba)3, BINAP and Sodium tert. butoxide in dioxane and heated to 100°C for 1 hr in a CEM discover microwave. Followed the workup process using dichloromethane and saturated aq. sodium bicarbonate solution crude compound was obtained which was purified using column chromatography (using 0%-10% methanol in dichloromethane as an eluent) to get the pure compound 36. This reported scheme has not discussed anything about the crystal nature of the molecule [12,13]. The detailed reaction pathway in Product patent for the synthesis of Ribociclib is Scheme 14.

Scheme 14: Drug discovery route for ribociclib.
Compound 34 was synthesised in seven chemical transformations and then coupled with 35 to achieve 36% yield. Use of highly expensive and corrosive reagents, low overall yield, lengthy steps, extensive purification techniques as column purification and long reaction time make the overall process cumbersome for commercialization. To establish commercialization process further process development activities were required.
Total synthesis of Ribociclib succinate (by novartis) (first process chemistry route)
Novartis also proposed the non-hydrate succinate salt of Ribociclib. The succinate salt had eminent features such as good stability, good solubility and non-hygroscopicity which improved the formulation process. As per this improved process not only the extensive column purification technique was removed also the yield for the synthesis of compound 34 increased to 30%. The reported process not only shortened the synthetic steps but also increased the overall yield from 0.9% to 12% [14]. This compound 37 exists in stable polymorphic form A (Scheme 15).

Scheme 15:Synthetic scheme by novartis for ribociclib succinate.
Second process chemistry route (WO2016192522A1)
Another process for the synthesis of intermediate 42 has been reported wherein N, N-dimethyl-2-carbonyl propenamide 38 is halogenated to get compound 39 as N, N-dimethyl-1-halo-2-carbonyl propanamide, that undergoes substitution reaction and reaction with malononitrile to obtain N, N-dimethyl-1,1-dicyano-3-carbonyl-butyramide 40. The compound 40 is subjected to cyclization to afford 2-methoxy-5-(N, N-dimethyl formamido)-3-pyrrylformonitrile 41, coupling of 40 with bromocyclopentane to get Ribociclib intermediate 42 as N-cylcopentyl- 2-methoxy-5-(N, N-dimethylformamido)-3-pyrrylformonitrile. The condensation between 42 and N-(5-(1-piperazino)-2-piperidyl) guanidine 30 results in Ribociclib. The reported process is viable for commercial production in terms of availability of raw materials, simple process technique, highly economical and environment friendly process. The entire chemical reaction is shown in Scheme 16.

Scheme 16:Synthesis of ribociclib using novel intermediate.
Polymorphism in Ribociclib
Ribociclib crystallizes in different polymorphic forms such as various crystalline forms and amorphous forms. By the reported synthetic methods, ribociclib so obtained contain an unacceptable high residual content that need to be eliminated and solid forms should comply as per ICH guidelines for residual solvents. As a free base the drug exists in different polymorphic forms as summarised in Table 8:
| Form | XRD (2θ values) | DSC (°C) |
|---|---|---|
| 1 | 12.5,12.6,13.3,13.6,14.8,18.1,18.3,21.8, 22.9,26.9±0.2. |
197.78 |
| 6 | 6.4,8.2, 11.3,15.3,16.6,17.7,19.1,20.2, 22.4±0.2. |
Two endotherms one at 193.09 and another at 197.85 |
| 7 | 5.3,10.7,11.0,13.0,13.9,14.7,15.9,20.8,21.1, 23.2±0.2. |
200.43 |
| 8 | 7.6,8.2,9.1,11.1,12.7,15.2,18.9,20.9,26.7, 28.2±0.2. |
Two endotherms one at 193.81 and another at 198.38 |
Table 8: The drug exists in different polymorphic forms as summarised.
As reported by Novartis, ribociclib succinate exhibit polymorphic form A, but as cited in other literatures, the mono-succinate salt exhibit crystalline transformation in various solvents and has poor reproducibility during process development studies [15]. Interestingly, the reported succinate salt is either hydrate or non-hydrate compound wherein the hydrate has poor solubility with 0.5 mg/ml and nonhydrate has better solubility.
Ribociclib is also available as hemi-succinate salt which is more stable than the reported prior art succinate salt. Other salts as reported is summarised in Table 9:
| Salt | XRD (2θ values) | DSC (°C) | TGA (wt. loss) |
|---|---|---|---|
| Hemi-succinate salt, crystalline Form A | 4.7,10.6,14.2,20.0,21.3,22.1, 23.9±0.2. |
Endothermic peak around 180°C | 12.5 % up-to 118°C |
| Mono-succinate crystalline Form-1 (anhydrous) | 6.83,7.82,11.24,11.88,12.46,13.06,15.7, 16.7,19.4,20.6,22.7,24.4,26.3±0.2. |
Endothermic peak around 197°C | 2.0 % up-to 178°C |
| Adipate crystalline Form A | 4.8,14.0,16.1,18.0,19.2,19.8,22.2, 24.9±0.2. |
Endothermic peak around 177°C | 2.1 % up-to 159°C |
| Maleate crystalline Form A | 8.5,16.5,17.1,21.9,24.5,29.3±0.2 | Endothermic peak around 207°C | 3.1 % up-to 138°C |
| Glycollate crystalline Form A | 10.1,12.4,13.3,16.8,19.5,21.3,21.8, 23.9±0.2. |
Endothermic peak at 253°C | 3.5 % up-to 176°C |
Table 9: Other salts which is more stable than the reported prior art succinate salt.
Ribociclib summary
The reported literature only defines limited process development schemes as the product is newly approved by USFDA. As per the cited references, none of the reported schemes has been commercialized. So, the scope of improvement in terms of viable, safe, economical, robust and efficient process is still in demand. As far as polymorphism is concerned, even though few stable polymorphs are reported but an intensive research is required for future references. At this stage literature is not sufficient to conclude the stable form and its application in drug product development. Therefore, through scientific understanding with critical analysis is required to have best possible route for commercial manufacturing with stable polymorphs.
Abemaciclib 54 an orally active agent developed by Eli Lilly pharmaceuticals, is still under investigation for breast cancer, NSCLC or pancreatic cancer. Based on its Phase-III clinical trials data, was approved by USFDA for treatment of certain breast cancers. It is also reported to exist as hydrochloride salt or as mesylate salt. Chemically, it is designated as N-(5-((4-ethylpiperazin-1-yl) methyl) pyridin-2-yl)-5- fluoro-4-(7-fluoro-2-methyl-3-propan-2-ylbenzimidazol-5-yl) pyrimidin- 2-amine, structurally as shown in Figure 4:

Figure 4: Abemaciclib.
Drug discovery route
The drug discovery route synthesised abemaciclib in eight sequential steps starting from 1-ethyl piperazine 43. This is subjected to reductive amination with 6-bromo-pyridine-3-carbaldehyde 44 leading to 45 which on substitution reaction with ammonia (using cuprous oxide as a reagent) affords 46. The 46 is designated as the one half chemical moiety of the parent compound. On the other hand, for the synthesis of other half of the parent compound isopropyl amine 49 is subjected to acylation using acetyl chloride leading to N-isopropyl acetamide 50, which upon reaction with 4-bromo-2,6-dilfuorophenylamine 48 affords 47. The entire reaction pathway is elaborated in Scheme 17.

Scheme 17:Drug discovery route for abemaciclib.
Intramolecular cyclization of 47 in the presence of N-methyl formamide at 70°C-75°C affords 51 which on hydroboration followed by palladium coupling with 4-bromo-2,6-difluorophenylamine leading to N-1 abemaciclib 53 (another half of the parent compound). Compound 53 is reacted with 46 to afford 54 as amorphous solid. The product patent also discloses its crystalline form as Form-I and Form-III.
Process chemistry route-I
In this approach a novel intermediate 55 was used for the synthesis of abemaciclib. Three chemical transformations starting from 58 leads to novel intermediate 55. In this reference, synthetic procedure for 58 starting from commercially available 2,6-difluoroaniline is also disclosed [16]. For the synthesis of 55, two different reaction pathways were designed and reported yield in both pathways were same, but the only difference lies in intramolecular cyclization (either early or late in the molecule) as shown in Scheme 18:

Scheme 18:1st Reaction pathway (late-cyclization strategy).
Late-cyclization strategy
Starting from 58 using DMF-DMA adduct compound 57 is obtained which on treatment with N-isopropyl acetamide leads to 56. The intramolecular cyclization of 56 using DMF and potassium tert. butoxide affords 55 (Scheme 19).

Scheme 19:2nd Reaction pathway (early-cyclization strategy).
Early-cyclization strategy
In this strategy 58 was treated with N-diisopropylacetamide in dioxane at 95°C-105°C to afford 59 in 84.6% yields. This was cyclized in the presence of 60% sodium hydride to achieve 60 in 83.3% yields which on treatment with DMF-DMA adduct leads to 55.
By using this novel intermediate 55, compound 54 was synthesised as per the reaction Scheme 20:

Scheme 20:Synthesis of abemaciclib using novel intermediate 55.
Process chemistry route-II
Short and concise route (in three steps) from easily accessible raw materials is reported in the literature utilizing Leuckart-Wallach reaction conditions for reductive amination step. As per the cited literature, chloride group in 2nd position of compound 52 is highly reactive in palladium catalysed reactions. As a result, this bond was targeted to undergo Suzuki coupling between 51a and 52. Use of PdCl2(PPh3)2 with sodium carbonate in dimethoxyethane at 80°C afforded compound 53 in 66% yield. For C-N bond formation Buchwald-Hartwig type coupling was studied wherein t-amyl alcohol as a solvent and DPEPhos as a catalyst was effective with 99% yield [17]. Finally, for reductive amination step, Leuckart-Wallach conditions wherein formic acid as reducing agent without base was employed. The water formed in the reaction was the major factor for in-complete reaction. Thus, trimethyl orthoformate was employed as water trapping agent to facilitate the forward reaction with isolated yield of 74%. Trimethyl orthoformate only works when it is used either as a solvent or co-solvent and facilitates the formation of imine.
The entire reaction pathway is shown in the Scheme 21.

Scheme 21:Synthesis of abemaciclib using leuckart-wallach reaction conditions.
Impurity reported in this approach
One of the major impurities reported in this approach is compound 64 (Figure 5), although this was observed during the process development studies but the reaction on higher scale need to be evaluated for the impurity generation [18-20].

Figure 5: Compound 64.
Polymorphism in Abemaciclib
The product patent also discloses different polymorphic forms of Abemaciclib as summarised in Table 10:
| Forms | Physio-chemical properties | XRD (2θ values) |
|---|---|---|
| Crystalline Form-I | Yellow solid, highly hygroscopic especially at high humidity and/or temperature; also have unfavourable electrostatic properties which significantly hamper the formulation development. | 4.51,5.89,8.98,11.20,12.57,13.09,15.93,16.31,17.01,18.58,18.82,20.86,21.90,23.12,23.53,26.71,26.85±0.2. |
| Crystalline Form-II | Yellow solid, increase solubility as compared to form-I but hygroscopicity is a problem especially at high humidity and/or temperature, also exhibited difficulty in the preparation of oral dosage forms. | 7.44,10.91,11.54,12.13,13.89,14.91,15.63,16.06,18.59,18.94,20.43,21.29,21.91,22.13,22.45,23.12,23.42,25.95,29.42±0.2. |
Table 10: The product patent also discloses different polymorphic forms of abemaciclib as summarised in table-10.
Abemaciclib form-IV
Abemaciclib form-I was suspended in acetonitrile (10V) and heated to reflux under stirring for 2 hrs, subsequently for 1 hr at 20°C. Filtered the slurry and dried under educed pressure for 2 hrs.
6.0,6.8,7.5,10.4,12.0,13.4,13.9,15.3,15.6,16.3,18.2,18.5,19.2,19.9,21.0,2 2.2,22.7,25.0,26.1,27.1,28.2,31.7 ± 0.2.
Endotherms (onset temperature): 123°C, 174°C, 181°C.
Water content
At ambient temperature it already contains 2.3% w/w water which increase to maximum of 3.4% at 95% RH representing 1 eq. of water molecule. Crystalline form remains unchanged after completion of all the experiments.
The crystalline form-IV provides the edge over prior art forms in terms of stability, reduced hygroscopicity, and superior solubility as compared to form-III.
As per the cited references, none of the reported schemes has been scaled up to a commercial level. So, the scope of improvement in terms of viable, safe, economical, robust, efficient process is still there. Since, the drug is still in trial phase so lot of scope in terms of process development study is present. As far as polymorphism is concerned, none of the reported polymorphs are good enough for drug product development therefore a vast and concrete research studies are required to reach a level of stable polymorphs for drug product development. A through scientific understanding with critical analysis is required to have best possible route for commercial manufacturing with stable polymorphs.
[PubMed]
[CrossRef] [Google Scholar] [PubMed]
[CrossRef] [Google Scholar] [PubMed]
[CrossRef] [Google Scholar] [PubMed]
Citation: Kumar P, Yaman SM, Mazlee MTF, Dhande P, Mhetre S (2022) Synthetic Review of Manufacturing Routes to CDK4/6 Inhibitor Drugs: Palbociclib, Ribociclib and Abemaciclib. Organic Chem Curr Res. 11:254.
Received: 21-Jan-2022, Manuscript No. OCCR-22-15542; Editor assigned: 24-Jan-2022, Pre QC No. OCCR-22-15542 (PQ); Reviewed: 07-Feb-2022, QC No. OCCR-22-15542; Revised: 12-Feb-2022, Manuscript No. OCCR-22-15542 (R); Published: 19-Feb-2022 , DOI: 10.35841/2161-0401.22.11.254
Copyright: © 2022 Kumar P, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.