Journal of Hematology & Thromboembolic Diseases
Open Access
ISSN: 2329-8790
ISSN: 2329-8790
Review Article - (2014) Volume 2, Issue 5
Multiple Myeloma (MM) is malignant haematological disease characterized by uncontrolled proliferation of monoclonal plasma cells (PC) in bone marrow (BM). The receptor, CXCR4 is widely expressed on hematopoietic cells including MM cells and respond to CXCL12 gradients for mobilization through blood stream and bone marrow. CXCL12 plays an important role in recruitment of MM cells to bone marrow microenvironment and formation of neoangiogenic niches supporting tumor growth, survival and metastasis. The integral role of this chemokine-receptor axis in development of MM makes it a desirable target for therapeutics. In this review, we outline the background on MM and role of specific chemokine CXCL12 in the disease with an attempt to highlight the targeted therapeutics for its signaling receptor CXCR4 in MM.
Keywords: Multiple myeloma; Hematopoietic cells; Bone marrow
Multiple Myeloma (MM) is malignant haematological disease characterized by uncontrolled proliferation of monoclonal plasma cells (PC) in bone marrow (BM). It is estimated that there are going to be 24,050 new cases of MM in the year 2014, this represents 1.4% of all new cancer cases. In current scenario the 5 year survival rate is 44.9%. While rates for new myeloma cases have been rising on average 0.7% each year (over 2002-2011), death rates have been falling on average 1.7% each year (over 2001-2010) owing to further insight gained into the biology and genetics of multiple myeloma as well as increased in therapeutic options improving patient outcomes. In this review, we outline the background on MM and role of CXCL12/CXCR4 chemokine receptor axis in the disease with an attempt to highlight the targeted therapeutics for its signaling receptor CXCR4 in MM.
The manifestations of MM include extensive skeletal destruction due to lytic bone lesions, loss of bone density leading to fractures, bone pain, hypercalcemia and renal insufficiency. The destruction of the skeletal tissue is the major cause of morbidity in MM patients [1].
The origin of MM lies in the development and differentiation of B cells. In the B cell differentiation and development process takes place in the BM where immunoglobulin (Ig) genes comprising variable (V), diversity (D) and joining (J) gene segments rearrange to generate the primary Ig repertoire. The assembly of a functional IgH-IgL complex also called pre B-cell receptor, BCR, on the surface of B cells allows them to escape apoptosis and migrate from BM environment to other lymphoid organs. When the virgin B cells reach the germinal center (GC) in the lymph node, the cells expressing a functional BCR undergo affinity maturation in response to antigen-presenting cells (APCs) through somatic hypermutation (SHM) and the class switch recombination (CSR) to produce antibodies of high specificity and avidity with different functional characteristics. Interestingly, the cells can still undergo maturation to a memory B cell and migrate if aberrant CSR occurs. These aberrant cells may additionally undergo oncogene deregulation allowing an acquired ability to survive and proliferate and further accumulate secondary hits. The resulting aberrant cells lead to emergence of malignant myeloma PC in the BM. These malignant PC continue their journey from lymph nodes across to the bone marrow microenvironment and localize in contact with stromal cells [2,3]. This localization of PC with BM stromal cells is critical first step in homing [4,5]. Once PCs are within the BM, they form specialized tumor niche that support plasma cells survival in a cellular environment that includes hematopoietic as well as non-hematopoietic cells like fibroblasts, stromal cells, endothelial cells (ECs), along with structural osteoclasts and osteoblasts. The non-cellular microenvironment components include the extracellular matrix (ECM) and the cytokines, chemokines and growth factors [6].
The direct interaction of MM cells with BM microenvironment cells activate signaling pathway mediating growth, survival, drug resistance, migration of MM cells [7], as well as osteoclastogenesis [8], angiogenesis and secretion of several chemokines, such as interleukin 6 (IL-6) [9], vascular endothelial growth factor (VEGF) [10], stromal cell-derived factor 1 (SDF-1) [11], and insulin-like growth factor (IGF1) [12]. PCs in the BM specifically secrete Angiopoietin-1, FGF-2, MMPs, TGF-β, TNFα and VEGF. The adhesion of MM cells to either BM stromal cells or ECM is mediated through adhesion molecules like CD44, very late antigen 4 (VLA-4), VLA-5, intracellular adhesion molecule (ICAM-1), NCAM, syndecan 1 and MPC-1. The cell-cell interactions mediated by adhesion molecules between PCs and BM cell initiate transcription and secretion of cytokines, such as CXCL12 (SDF-1), IGF-1, IL-6, MCP-1 (CCL2), and VEGF by BM cells [13].
It is now recognized that major factor contributing to migration of plasma cells into and out of bone marrow, as well as their chemotaxis within the bone marrow microenvironment, is directed by the interaction of chemokines and their receptors [14]. Of these, CXCL12 and its receptor, CXCR4 and CXCR7 play a fundamental role in MM pathogenesis, because they mediate the MM cell homing to the BM. In this review we will focus on the role of CXCL12 /CXCR4 axis in MM and current and future strategies to target this axis leading to better clinical outcomes.
Chemokines are important for normal cell positioning and path finding during development, cellular activation and homeostatic cell trafficking under physiological conditions. A variety of cell types secrete chemokines either constitutively or after induction by stimuli like tissue damage or an infection [15,16]. Chemokines have long been associated with cancer [17] and have been shown to play multiple roles ranging from the recruitment and positioning of leukocytes, growth, invasion and metastasis of transformed cells, angiogenesis, modification of extracellular matrix, and organ specific metastasis.
There is extensive literature available on chemokines and their structure. The chemokine family comprises about 46 human ligands that signal by binding to specific receptors. Most chemokines are small (8–14 KDa) proteins that interact with G-protein coupled receptors [18].
Chemokines are divided into two major and two minor groups according to their structure, arrangement and spacing of two conserved cysteine residues. The two major groups are CC- and CXC- chemokines, and the two minor groups are CX3C and XCs. The CXC family has a non-conserved intervening amino acid residue in between the two cysteine residues; the CC family has two cysteine residues in juxtaposition, whereas in the CX3C family there are three intervening amino acids in between the two cysteine residues. The CC chemokines induce migration of monocytes, lymphocytes, basophils, eosinophils and dendritic cells. The CXC family can be further subdivided into two categories based on the presence of the sequence glutamic acid-leucine-arginine (or ELR) at the NH2 terminus immediately before the first cysteine of the CXC motif [19]. The ELR motif dictates chemokine specificity for binding to their related receptors. ELR+ CXC chemokines possess angiogenic and neutrophil chemotactic properties whereas most ELR- CXC chemokines are angiostatic and attract lymphocytes and natural killer cells. Similar to chemokines, chemokine receptors adopt a matching nomenclature, for example the chemokine fractalkine or CX3CL1 (L for ligand) binds to CX3CR1 (R for receptor) [20,21]. In addition, analyses of chemokine ligands and their receptors families are classifiable based on patterns of genomic organization attributable to evolutionary pressures [22].
Based on their expression patterns and functions, chemokines can also be grouped as: inflammatory (e.g. CCL1-13, CCL23, 24, CXCL1-3, CXCL5-11), involved in leukocyte mobilization during inflammation or homeostatic (e.g. CCL14-16, CCL25, CCL27, CXCL12, 13) [20,21] implying involvement in leukocyte trafficking during normal physiological states. Of these CXCL12 is a homeostatic chemokine with major function to regulate hematopoietic cell trafficking with in bone marrow and secondary lymphoid tissue. The abrogation of CXCL12/CXCR4 pathway function in CXCL12 knockout studies show deregulated bone marrow colonization by hematopoetic cells. In adults, the retention and homing of stromal cells to the bone marrow microenvironment and lymphocyte trafficking is controlled by CXCL12/CXCR4 pathway. CXCL12 is constitutively expressed in several organs including bone marrow, brain, heart, lung, liver, muscle, kidney, skeletal and skin tissue. CXCL12 secretion is also associated with tissue damage induced by infarction, ischemia, liver damage, bleeding, irradiation, and chemotherapy as well as at tumour sites in lymphoma, glioma, ovarian and pancreatic cancer, and at sites of metastasis in breast, thyroid cancer, neuroblastoma and haematological malignancies [22-28].
Chemokine receptors
Chemokine receptors belong to class A seven transmembrane G-protein-coupled receptors and consist of 350 amino acids on average. Primary receptors are defined as CXCR, CCR, CR or CX3CR [29,30]. These include six CXC receptors (CXCR1-6), eleven CC receptors (CCR1-11), one CX3C (CX3CR1) and one C receptor (XCR1). The chemokine receptors are grouped according to the ligands they bind, i.e., CCR1-10, CXCR1-6, CX3CR1, and XCR1. There are 18 functionally signalling chemokine receptors and four atypical chemokine receptors acknowledged in the human proteome. Atypical receptors include Duffy Antigen Receptor for Chemokines (DARC), D6, CCX-CKR and CXCR7, which bind chemokines with high affinity but are unable to activate the signal transduction pathways [28,29,31,32]. The function of atypical chemokine receptors is to modulate immune responses by scavenging, sequestration, buffering as well as intracellular transport of chemokines from inflammatory sites [28,30,33-35].
The chemokine system is highly promiscuous. Several chemokines can bind to the same receptor and vice versa, although some receptor–ligand interactions are highly specific and selective, e.g., CCR9–CCL25 and CXCR6-CXCL16. This promiscuous interaction pattern together with the large number of receptors and ligands enables the chemokine system to propagate a great variety of cell functions [29,36-39]. Chemokines in addition to binding to their receptors also bind to glycosaminoglycans (GAGs) present in ECM and found on leukocytes, epithelial and endothelial cells [29,37,40]. Chemokines bind to GAGs leading to establishment of a local chemokine concentration gradient and thus GAGs assist in the attraction of cells expressing the specific chemokine receptors [29,40]. In addition, GAGs may protect chemokines from proteolysis and degradation and thereby allowing the possibility of chemokine oligomerization [40].
The receptor CXCR4 is expressed on almost all of the hematopoietic cells, embryonic pluripotent and tissue-committed stem cells, allowing them to migrate and invade along CXCL12 gradients [15,41-44]. Specifically in malignant cells the chemokine receptor that is most commonly found is the receptor CXCR4. At least 23 different types of tumour cells from human cancers of epithelial, mesenchymal and haematopoietic origin express CXCR4 [45]. In cancer, CXCL12 plays a role in the mobilization and recruitment of these cells to the inflammatory tumor microenvironment, neo-angiogenic niches, supporting revascularization, tumor growth and metastasis [46].
The role of CXCL12/CXCR4 in cancer progression is one of the best delineated chemokine–receptor interactions in malignant cells. The initial studies to understand the functioning of this axis established that CXCL12 bound exclusively to CXCR4 and that CXCR4 was its sole receptor, however in the year 2005, CXCR7 was identified as an additional receptor for CXCL12 [47]. CXCR7 is regarded as a non-classic scavenging or decoy receptor that modulates the function of CXCR4. It has been demonstrated that CXCR4 and CXCR7 form a receptor unit for successful CXCL12 signaling in some cells [48]. Moreover, signal transduction can occur through CXCR7 (instead of CXCR4) in specific cells [49].
Major biological functions of chemokines and receptors
Chemokines are of great importance in regulating the physiology and pathology by controlling directional movement of cells [15,50]. The activity of chemokines is initiated by binding of chemokine to its specific G-protein coupled receptor. In a two-step model the first step constitutes recognition and binding of chemokine to the receptor followed by conformational change in the chemokine at N terminus which then can act as a guanine nucleotide exchange factor leading to exchange of bound GDP for GTP in α subunit of the G proteins [51]. The G protein then dissociates from the receptor and activates several effector molecules downstream initiating a molecular signaling cascade in the cytoplasm. Depending on the effector molecules the signaling cascades result in diverse physiological processes including migration and trafficking of leukocytes, degranulation of leukocytes, cell differentiation and angiogenesis thus affecting development and response of the immune system including migration of dendritic cells, extravasation of leukocytes and maturation of B- and T- cells.
The functions of chemokines make them critical players in pathological processes characterized by chronic inflammation including microbial infections, auto-immune diseases, tumor microenvironment, metastasis and angiogenesis [50,52]. Specifically in case of cancer, chemokines and receptors are abundantly expressed in chronic inflammatory conditions that occur downstream of genetic events contributing to neoplastic transformation and affect multiple pathways of tumor progression and predispose to cancer [15,50]. Activation of chemokine receptors initiates signaling cascades that may stimulate neoplastic transformation and further contribute to providing directional cues for migration/metastasis, shaping the tumor microenvironment, and providing survival and/or growth signals.
As mentioned earlier, tumor cells respond to chemokines in their microenvironment based on expression of chemokine receptors. In case of Kaposi’s sarcoma, a widely found neoplasm in AIDS patients, KSHV-GPCR, human G-protein coupled receptor encoded by Kaposi's sarcoma herpes virus shares a high degree of homology with human CXCR2 [53]. This receptor, when expressed on infected cells, triggers a constitutive signal which is further up-regulated by binding of CXCL8 and CXCL1 [54]. This interaction leads to molecular signaling cascade causing neoplastic cellular transformation [55]. In numerous types of cancer, the malignant cells exhibit increased or aberrant expression of particular chemokine receptors relative to their normal counterparts, notably CXCR4, CCR7, and CCR10 [45,56-58].
The tumor microenvironment exists in a state of chronic inflammation characterized by the presence of malignant cells, aberrant vascular network and abundance of inflammatory mediators. In such an environment, chemokines and their receptors play roles in modulating angiogenesis [59], cell recruitment [45], tumor survival [60] and proliferation [61-63], and lead to progression of the cancer. The chemokines play significant role in directing organ-specific metastasis; the chemokine receptor expression patterns on cancer cells and the localization of the corresponding ligands provide clues for directional metastasis [28]. Breast cancer for example, has a tendency to metastasize to lymph node, bone marrow, lung and liver. The metastatic process is influenced by chemokine network by directing the migration of receptor-bearing tumor cells to sites of metastases where the ligands are expressed, similar to the process of lymphocyte trafficking [28].
Chemokines and their receptors play an essential role in the regulation of B-lymphocyte trafficking. MM being a disorder of B- cells, chemokines are integral to its growth and progression. As mentioned earlier in this article, the lymphocytes undergo series of changes during different steps of their development, i.e. maturation, differentiation and activation. Incidentally, this also includes the possibility of alteration in expression of chemokine receptor and thus influencing the migration in response to different sets of chemokines [64]. In normal conditions the differentiated B-cells or PC, are destined to lodge in different tissues, i.e. spleen, lymph node and BM and do not recirculate. However, the transition of normal plasma cells to monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple Myeloma (SMM) to MM consists of many oncogenic events [65]. The dysregulation of cyclin D gene, mutations of NRAS and KRAS, constitutive activation of the nuclear factor κB (NFκB) and secondary translocations in an Ig locus are the few events that contribute to oncogenicity. The survival of these transformed cells is dependent on the bone marrow microenvironment with further demand on these cells to express distinctive chemokine receptor profiles at the site of disease activity [65-69]. Pellegrino et al have shown that in comparison to healthy endothelial cells, the bone marrow endothelial cells from patients with MM were found to express and secrete higher amounts of the CXC chemokines CXCL8, CXCL11, CXCL12, and CCL2 [70]. Pellegrino et al further showed that paired plasma cells and several MM cell lines express cognate receptors of each chemokine [70].
The process of homing is described as the migration of transformed PC through blood to the bone marrow niches which requires active navigation with the first step being rolling of the MM cells along the EC through selectin followed by adhesion and extravasation by activation of LFA-1 and VLA-4 integrins expressed by MM cells [71]. The events that make MM cell migration different from their B-cell precursors include increased sensitivity to the CXCL12 and the down-regulation of CXCR5 and CCR7 [72,73]. In addition, MM cells display a complex expression pattern of chemokine receptors including CCR1, CXCR3, CXCR4 and CCR6 on MM cell lines. This was confirmed by Nakayama et al as they found CXCR4, CXCR6, CCR10 and CCR3 on fresh myeloma plasma cells [17,41,74].
The CXCL12/CXCR4 axis is crucial for normal development as demonstrated by the fact that targeted disruption of either CXCR4 or CXCL12 is lethal in mice, resulting in failure of hematopoietic stem-cell (HSC) migration from liver to bone marrow with defects in B-lymphopoeisis, and cerebellar dysgenesis [75-77].
The CXCL12/CXCR4 pathway is responsible for retention in the BM of acute lymphoid leukemia (ALL), acute myeloid leukemia (AML) and multiple myeloma (MM) cells [78,79]. This pathway a key regulator of MM cell homing, adhesion and motility and extravasation of tumor cells in bone (Figure 1) [80,81]. Large variations in CXCR4 expression have been observed which range from 10 to 100% on MM cells [81]. A CXCR4 knockdown leads to significant inhibition of migration of MM cell lines and primary CD138+ cells towards CXCL12 [81]. While myeloma PCs and endothelium express CXCR4, bone marrow stromal cells express ligand CXCL12 for this receptor [16,82]. Thus migration of PCs across the endothelium lining of the bone marrow sinuses leads to homing and localization of these myeloma PCs near stromal cells demonstrating a positive correlation of the gradient of CXCL12 protein towards chemotactic activity of myeloma cells through integrins. An increased expression of CXCR4 is associated with a poor prognosis and is associated with advanced and metastatic disease [83].
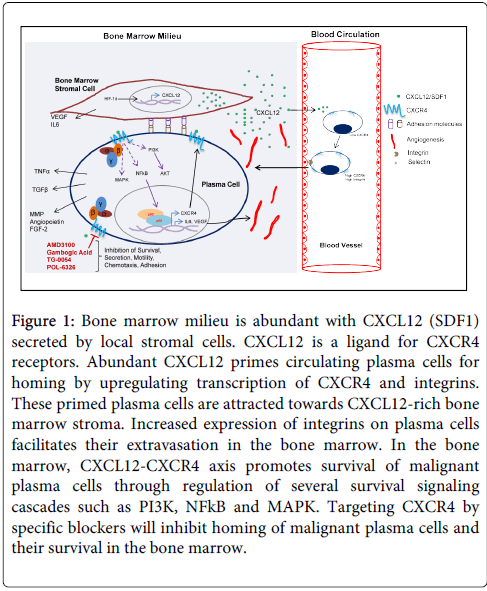
Figure 1: Bone marrow milieu is abundant with CXCL12 (SDF1) secreted by local stromal cells. CXCL12 is a ligand for CXCR4 receptors. Abundant CXCL12 primes circulating plasma cells for homing by upregulating transcription of CXCR4 and integrins. These primed plasma cells are attracted towards CXCL12-rich bone marrow stroma. Increased expression of integrins on plasma cells facilitates their extravasation in the bone marrow. In the bone marrow, CXCL12-CXCR4 axis promotes survival of malignant plasma cells through regulation of several survival signaling cascades such as PI3K, NFkB and MAPK. Targeting CXCR4 by specific blockers will inhibit homing of malignant plasma cells and their survival in the bone marrow.
The exposure of MM cells to chemokines CXCL8/IL-8 and CXCL12/SDF-1a result in enhanced cellular proliferation and chemotaxis [70]. Further, CXCL12/CXCR4 axis promotes migration of myeloma cells by transient upregulation of integrin dimer VLA-4 (α4β1) on cell surface, inducing cell adhesion to the VCAM-1 receptor on endothelium, and contributing to the trafficking of myeloma cells in the bone marrow microenvironment and their localization in close proximity to stromal cells, forming tumor niche [71,84]. CXCL12 has been further implicated in homing and localization of MM cells in the bone marrow through upregulation of its own secretion by stromal cells, promoting greater expression of integrins and hence contributing to enhanced homing. The CXCL12 expressed by stromal cells induces proliferation of MM cells and induces phosphorylation of mitogen-activated protein kinase kinase 1/2 (MEK1/2), p42/44 mitogen-activated protein kinase (MAPK), and Akt in a time-dependent fashion leading to cell migration [7]. Additionally, CXCL12 induces interleukin-6 (IL-6) and vascular endothelial growth factor (VEGF) secretion, indicating contribution to the growth of MM [7]. The role of this axis in survival is evident from induction of NF-κB in MM cells promoting anti-apoptotic pathways [7,85] as well as protection from dexamethasone-induced apoptosis through activation of the mitogen-activated protein (MAP) Akt pathway, implying a role in drug resistance.
Disruption of CXCL12/CXCR4 also plays a key role in chemotherapy-based extravasation of MM cells and progenitor cells from BM to peripheral blood as well as mobilization of myeloma cells on decrease of levels of CXCL12 in serum and reduced expression of CXCR4 on surface of these cells [81,86].
The hypoxic environment in BM is an important factor for the evolution of tumor cells. Increased levels of CXCL12 have been observed in distinct niches of hypoxia along with colocalization of progenitor cells in these niches, suggesting that HIF-1-regulated CXCL12 expression plays an important role within the normal bone marrow microenvironment. In addition to inducing CXCL12, HIF-1 enhances the expression and function of CXCR4 on normal and malignant cells. Hypoxia-inducible transcription factor, HIF -1α is widely expressed throughout the bone marrow, while HIF-2α is restricted to macrophages and CD138+cells. It has been shown that HIF-induces CXCL-12 leading to malignant transformation of MM cells [87,88].
To summarize, the CXCL12/CXCR4 pathway may be critical for myeloma progression via multiple mechanisms: (i) by promoting cancer cell growth, survival, migration and invasion. This is evident in mobilization or egress of cells out of the bone marrow which could be enhanced by disrupting the CXCL12/CXCR4 axis as well as indirect role of CXCL12 in pathogenic responses that allow tumor survival [89] (ii) by recruiting “distal stroma” to indirectly facilitate homing, tumor growth and metastasis; and (iii) by promoting angiogenesis to support tumor growth [89]. As evident from the existing studies, a continuous circulation of the MM cells in the peripheral blood and homing back to the BM is critical for the progression of MM. This continuous trafficking (homing-egress) allows the progression or “metastasis” of the disease to new BM sites [90].
As discussed earlier, the interaction of MM cells with ECM proteins and BM cells, as well as factors in the BM microenvironment (cytokines, chemokines, angiogenesis), plays a crucial role in MM pathogenesis and drug resistance [7,91,92]. MM remains incurable because most patients will eventually relapse or become refractory to the treatments. Although the treatments have improved, the major problem in MM is the resistance to therapy. The host immune system and the microenvironment, proliferation, survival, migration, and resistance to drugs are responsible for some clinical manifestations of MM and provide protection to PC from cytotoxic effect of chemo- and radiotherapy.
The mechanisms contributing to resistance to therapy include intrinsic genetic mechanisms (mutated p53), acquired resistance following exposure to conventional chemotherapeutic treatment (overexpression of P-glycoprotein) or by binding of multiple myeloma cells to extracellular matrix proteins induces cell-adhesion-mediated drug resistance (CAM-DR) to conventional chemotherapy (through upregulation of p27, NFκB). The immune system and bone microenvironment and secretion of growth factors (TGF-β) further induce transcription and secretion of cytokines, which in turn confer drug resistance (interleukin 6 secretion by BMSCs, osteoclasts and endothelial cells protects from apoptosis by dexamethasone).
Novel agents are currently in development for the treatment MM, including immunomodulatory drugs, proteasome inhibitors, monoclonal antibodies, cell signaling targeted therapies, and strategies targeting the tumor microenvironment. Recent advances in the treatment of MM with novel therapeutic immunomodulatory agents such as thalidomide, pomalidomide, and lenalidomide that target the MM cells as well as the bone marrow microenvironment have induced significant responses [93]. However, many patients do not respond to these agents, indicating that further advances are required [93]. Current studies have focused on developing therapies that induce apoptosis of MM cells, even in the presence of the BM milieu, but even these have had limited success [94]. Other novel therapies being evaluated for MM intervention include proteasome inhibitors (bortezomib, carfilzomib, marizomib and ixazomib citrate), monoclonal antibodies (elotuzumab, siltuximab, daratumumab and BT-062), and drugs affecting an interaction with the tumor microenvironment (anti-VLA4 monoclonal antibody, chemokine CXCR4 inhibitor AMD-3100 and selectin inhibitor GMI-1070) [95]. The role of molecular targeted therapies is gaining focus in the treatment of relapsed/refractory multiple myeloma, including cell signaling targeted therapies (HDAC, PI3K/AKT/mTOR, p38 MAPK, Hsp90, Wnt, Notch, Hedgehog, and cell cycle) and strategies targeting the tumor microenvironment (hypoxia, angiogenesis, integrins, CD44, CXCR4, and selectins) [96]. Table 1 lists the prominent therapeutics currently in use as well as in clinical trials.
| Therapeutic Agents | Target | Clinical studies |
| Immunomodulatory agents: | ||
| Pomalidomide | Aantiangiogenic and immunomodulatory activities; Caspase-8 mediated apoptosis; | Approved by FDA in 2013 as a treatment for relapsed and refractory MM |
| Thalidomide | Decreased binding of tumor cells to BM stromal cells; | Numerous clinical trials have confirmed its efficacy, most often used in combination with other treatments |
| Lenalidomide | Inhibit secretion of cytokines from the BM, and stimulate immunity against myeloma cells | Approved by FDA in 2005 as a treatment for relapsed and refractory MM |
| Proteasome inhibitors: | ||
| Bortezomib | proteasome inhibitor- received accelerated FDA approval | Newly-diagnosed disease with dexamethasone: In combination liposomal doxorubicin; thalidomide; lenalidomide- (RVD) |
| Carfilzomib | selective proteasome inhibitor irreversibly binds to and inhibits the chymotrypsin-like activity of the 20S proteasome | Phase III confirmatory clinical trial, known as the ASPIRE trial, comparing carfilzomib, lenalidomide and dexamethasone versus lenalidomide and dexamethasone in patients with relapsed MM |
| Oprozomib | oral proteasome inhibitor to selectively target the proteasome | Phase 1b/2 study for treatment of MM |
| Ixazomib (MLN9708) | First oral proteasome inhibitor | Patients With Newly Diagnosed MM and Relapsed MM that is Not refractory to Bortezomib in combination Ixazomib+Lenalidomide+Dexamethasone |
| NPI-0052 | Second generation proteasome inhibitor that prevents the breakdown of proteins involved in signal transduction which blocks growth and survival in cancer cells | Phase 1/2, open-label in patients undergone prior bone marrow transplantation, with relapsed or relapsed/refractory MM |
| PR-924 | LMP‑7 immunoproteasome subunit | |
| MLN4924 | NEDD8-activating-enzyme: neddylation pathway upstream of the 20S proteasome; disrupts NF-κB activation | |
| HDAC Inhibitors: | ||
| Ricolinostat or ACY-1215 | selective HDAC6 inhibitor | Phase 1b studies in combination with bortezomib or lenalidomide in relapsed or refractory MM |
| Vorinostat | oral histone deacetylase (HDAC) inhibitor | Phase III vorinostat and bortezomib with relapsed and/or refractory MM |
| Panobinostat | Nonselective HDAC inhibitor | Phase II study of panobinostat plus bortezomib plus dexamethasone |
| in patients with MM | ||
| Monoclonal antibodies: | ||
| Elotuzumab- anti CS-1 MoAb | CS-1 mediated tumor cell adhesion and tumor growth via interaction with BM stromal cells | Phase 2, evaluated elotuzumab in combination with lenalidomide and low-dose dexamethasone in previously-treated MM patients |
| BT062 | CD138mAb conjugated to maytansinoid DM4 | Phase I/II trials, preliminary evidence of clinical benefit has been observed |
| Dacetuzumab | anti-CD40 Mo Ab, Downregulation of IL-6 receptor, and inducing death in MM cells |
|
| Lucatumumab human anti-CD40 MoAb | inhibits MM cell growth | Phase I study in patients with relapsed/refractory MM |
| Siltuximab- anti–IL-6 chimeric MoAb | Interleukin-6 | One trial with siltuximab and bortezomib in combination |
| Daratumumab- CD38 MoAb | CD38 mediated adhesion, signal transduction, and regulation of intracellular calcium |
Phase I/II study, single agent and in combination with lenalidomide and dexamethasone in relapsed and refractory MM |
| Bevacizumab- Anti-VEGF | Approved by FDA for other indications; to target upregulated VEGF in MM | Several trials in combination with either bortezomib, lenalidomide, or thalidomide |
| with low-dose dexamethasone | ||
| Denosumab | Receptor activator of nuclear factor-κB ligand (RANKL) inhibitor- the key mediator of osteoclastogenesis | Phase III registration trial, denosumab was noninferior to zoledronic acid in preventing or delaying the first on-study skeletal related events (SREs) |
| Kinase Inhibitors: | ||
| P38MAPK inhibitor | HSP27 expression | |
| Temsirolimus | mTOR | Phase I studies of lenalidomide |
| and temsirolimus (CCI-779) in patients with relapsing myeloma | ||
| Everolimus or RAD001 | mTOR | Everolimus in combination with lenalidomide patients with relapsed and refractory myeloma received |
| INK128, AZD 8055 | mTORC1/2 | |
| NVP-BEZ235 | mTORC1/2 and PI3 Kinase | |
| Afuresertib | Novel AKT inhibitor | Phase I/II study of afuresertib, bortezomib, and dexamethasone in patients with relapsed/ refractory myeloma |
| Perifosine | Oral Akt inhibitor | Phase I/II study, in combination with bortezomib with or without dexamethasone in patients with relapsed and refractory myeloma. |
| Accessory cells and cytokines | ||
| Bisphosphonates: Zolderonic acid Clodronic acid |
Inhibit osteoclast-mediated bone resorption and are indicated for the treatment of bone metastasis, reduce the risk of skeletal events, used as supportive therapy MM | Several clinical studies; Proapoptotic effects on cancer cells, cytotoxic synergy with chemotherapeutic agents, antiangiogenesis, interference with adhesion of cancer cells, and stimulation of host anticancer immune responses |
| Other agents | ||
| Ibrutinib | Btk inhibitor, Osteoclast formation and growth, cytotoxic to myeloma and enhances bortezomib and lenalidomide activities through NF-κB | orally available and irreversibly binds to Btk with excellent pharmacodynamics Phase I/II trials ongoing |
| ACE-011 (Sotatercept) | Activin A and ostoclastogenesis, a protein therapeutic based on the activin receptor type IIA, bone-forming agent | Anabolic effect in MM patients receiving standard chemoptherapy in Phase II |
| Homodeoxyharringtonine HHT alkaloid | PI3/AKT , JAK/STAT and HIF pathway | Approved in October 2012 by the US FDA for chronic myeloid leukemia; cytotoxicity in dexamethasone sensitive/resistant, additive antitumor effect with bortezomib in MM |
| Aplidine or didehydrodidemnin B | Anti-MM effect associated with suppression of a constellation of proliferative/antiapoptotic genes | Phase III clinical trials in Combination With Bortezomib and Dexamethasone in Patients With Relapsed and/or Refractory MM |
| JQ1 | BET Bromodomains, depletion of c-Myconcoprotein leading to cell cycle arrest and senescence | Preclinical studies |
Table 1: Clinical strategies for treatment of multiple myeloma approved or under clinical development.
The results of pre-clinical and clinical studies have revealed that the chemokine system represents a valuable target for the development of novel therapeutic strategies. The clinical trials evaluating chemokine receptor antagonists have gained significant focus as these are one of the most amenable drug targets in the huge battery of molecules that regulate inflammation and immunity [97].
Since activation of CXCL12/CXCR4 pathway has been found to be critical for tumor progression via multiple complementary mechanisms a keen focus has developed on targeting this pathway in solid tumors as well as in myeloma and leukemia [98,99]. The recent efforts in this area have been reviewed by Nigris et al, 2012 [93]. The agents currently being developed range from CXCR4 neutralizing antibodies, small synthetic or natural molecules, peptides and peptidomimetics to RNA interference. Specifically for the treatment of MM the ongoing clinical studies include agents AMD3100, modified peptide BKT140, Polyphemusin II derivative POL6326 and quinolone TG0054.
The approval of the first CXCR4 antagonist by FDA is plerixafor or AMD3100 for the mobilization of hematopoietic stem cells [100-103]. AMD3100 is a bicyclam derivative which reversibly competes with and inhibits CXCL12 binding to CXCR4 [Figure 1] [104,105]. An observation of leukocytosis in CD34 cells during clinical studies testing AMD3100 as an agent for treatment of human immunodeficiency virus (HIV) infection was made which led to categorization of this compound as a competitor to CXCL12 [106]. AMD3100 inhibited migration in vitro and homing of MM cells in vivo, as well as downstream signaling through the phosphatidylinositol 3-kinase (PI3K)/Akt and extracellular signal-regulated kinase (ERK) signaling pathways highlighting its potential in CXCL12 and CXCR4 mediated homing of MM cells to the bone marrow [81]. Previous studies have shown that CXCR4 inhibitor AMD3100 induces significant mobilization of hematopenic stem cells (HSCs) into the peripheral blood [107]. The MM cells can be mobilized into the circulation with the use of AMD3100 rendering them more sensitive to therapeutic agents by disrupting their adhesion to the BM microenvironment [79].
TG-0054 is an injectable small molecule, quinolone derivative that is selective CXCR4 antagonist with therapeutic potential. It has demonstrated excellent safety and PK/PD data in phase I and has entered phase II for stem cell transplantation in clinical trials for its role in stem cell mobilization in patients with multiple myeloma, non-Hodgkin lymphoma or Hodgkin disease (http//clinicaltrials.gov).
Polyphemusin II derivative POL6326 is a reversible competitive CXCR4 antagonist that blocks the interaction with the ligand CXCL12 resulting in the mobilization of stem cells from the bone marrow into the circulating blood. Phase I clinical trials have been successfully completed and currently POL6326 is being investigated as a stand-alone therapy in a Phase II clinical trial for its efficacy in autologous transplantation of hematopoietic stem cells in multiple myeloma patients (http//clinicaltrials.gov) [108].
The challenges in clinical translation of these agents due to poor pharmacological profile, has led to exploration of modulators of CXCR4 that are bioavailable, less toxic, effective and inhibit development of tumors in animal models. Meng et al have demonstrated that homoharringtonine (HHT), isolated from the Chinese evergreen Cephalotaxus harringtonia, induces significant cytotoxicity in dexamethasone-sensitive and -resistant and chemotherapy-sensitive MM cell lines as well as in chemotherapy resistant patient's myeloma cells, independent of interleukin-6 [109]. The mechanism of HHT cytotoxicity is related to down-regulation of Akt phosphorylation/activation and various target genes of Akt including nuclear factor kappa B, XIAP, cIAP and cyclin D1. The data from our lab has shown one such compound Gambogic acid, a xanthone derived from the resin of Garcinia hanburyi has 20 times more potent inhibitory effects on CXCR4 than AMD 3100 in vitro [110]. Furthermore, GA down-regulates expression of CXCR4 in MM cells.
Overall, more than 20 different chemical classes have been identified as CXCR4 antagonists, as reviewed comprehensively by Debnath et al, however, only a few have been studied in detail [111]. The major classes include tetrahydroquinolines, N-substituted indoles, 1,4-phenylenebis (methylene) derivatives, and N-containing heterocycles which are at different stages of evaluation and have unique features to make them desirable for different indications having implications of CXCL12/CXCR4 axis induced signaling. Novel therapeutics targeting CXCR4 in clinical trials along with their ClinicalTrials.gov Identifier are listed in Table 2. The main intent in clinical development of novel CXCR4 antagonists is being orally active and available, pharmacologically effective and safe, and synergistic in activity when used in combination therapy since these are meant for long term treatment.
| Compound | Company | Stage of development |
| AMD3100 | AnorMED | Glioma, Acute Myeloid Leukemia, Chronic Lymphocytic Leukemia |
| Genzyme | ||
| AMD11070 | Genzyme | Preclinical for hematological malignancies; Phase I in HIV-infected Patients Carrying X4-tropic Virus |
| NCT00361101 | ||
| TG-0054 | TaiGen Biotechnology Co. | Phase II, Hematopoietic stem cell mobilization in patients with MM, non-Hodgkin lymphoma or Hodgkin disease |
| NCT02104427 | ||
| POL6326 | Polyphor Ltd. | Phase II, Hematopoetic stem cell mobilization in patients with Leukemia, Lymphoma and Multiple Myeloma |
| NCT01413568 | ||
| T140 peptide | Kyoto University | Preclinical studies in hematological malignancies |
| KRH-2731, -3140, -3955 | Kureha | Preclinical development, orally available |
| BKT140 | Biokine Therapeutics Ltd. | Phase (I/II) MM |
| NCT01010880 | ||
| BL-8040 | Sheba Medical Center | Phase I/II, Relapsed AML, CML and Selected B-cell Malignancies with Imatinib |
| NCT02073019, NCT01838395, NCT02115672 | Bioline Rx | |
| BMS-936564 (MDX-1338) NCT01120457 | Bristol-Myers Squibb | Relapsed Acute Myelogenous Leukemia and Selected B-cell Malignancies |
| BMS-936564 | Bristol-Myers Squibb | Phase 1, AML and B cell cancers |
| NCT01120457 | (Anti-CXCR4) | |
| ALX-0651 | Ablynx | Phase I Proof of concept Nanobody Inhibiting CXCR4 |
| NCT01374503 |
Table 2: CXCR4 antagonists in clinical trials for treating hematological malignancies
Small molecules were favored in MM therapeutics directed towards CXCL12/CXCR4 axis initially however there are several challenges with respect to their accessibility to target site which is compounded by broad expression of CXCR4 with in the body leading to unwanted side effects. Small peptides on the other hand can be locally administered but their poor selectivity has led to clinical trial failures. In addition, targeting CXCR4 may not be sufficient as CXCL12 can bind to CXCR7 on cancer or stromal cells and induce signaling cascades. And last but not the least, anti CXCR4 therapy alone is not sufficient and requires to be dosed with chemo- or radiotherapy as currently being pursued in ongoing clinical trials.
There are also challenges associated with long term therapy conceptually as CXCR4 antagonists while inducing mobilization of hematological or metastatic cancer cells, may also mobilize normal stem cells leading to increased toxicity of chemo and radiotherapy and affect angiogenesis by recruiting endothelial precursor cells. Overcoming these challenges and better understanding of the biology as well identification of better agents would increase the chances of realizing the promise of CXCL12/CXCR4 pathway inhibition as a potentially effective strategy in MM treatment.
This work was partially supported by a grant from the International Myeloma Foundation (M.K.P.), and partly from Penn State Cancer Institute, Hershey, PA (S.G.A.). We apologize to all colleagues whose work could not be cited owing to space restrictions.