Journal of Clinical Trials
Open Access
ISSN: 2167-0870
ISSN: 2167-0870
Perspective - (2014) Volume 4, Issue 3
Background: Most critically ill patients are unable to comprehend or communicate and Substitute Decision Makers (SDMs) typically provide proxy consent for research. Ontario legislation requires individuals within the ‘circle of care’ to introduce research personnel to SDMs with some Research Ethics Boards (REBs) mandating physician involvement. We aim to conduct mixed methods, pilot Randomized Controlled Trial (RCT) comparing two strategies for introducing research to SDMs and a fully nested qualitative study evaluating SDM’s experience in being approached for consent.
Methods/Design: A multicentre, pilot, mixed methods RCT comparing different strategies [physician (MD) introduction vs. non-physician (non-MD) introduction] for initially introducing research personnel and research participation to SDMs (target n=150) of critically ill adults. In the intervention arm, physicians will introduce research coordinators (RCs) and study participation to SDMs using a standardized script. In the control arm, RCs will introduce themselves or be introduced by a non-MD member of the ICU team. We will consider the trial feasible if (i) ≤ 15% of physician introductions are missed due to lack of
physician availability (MD introduction arm) and (ii) cross-overs (from one arm to the other) occur in ≤15% of encounters. We expect that ≤20% of introductions will be missed due to inability to contact an SDM and moderate to high SDM questionnaire completion rates. In the qualitative study, we will interview 12 SDMs (6 MD and 6 non-MD introductions) to describe their experience in being approached.
Discussion: The Approach Trial will evaluate how to best approach SDMs to make encounters more comfortable, credible, informed, and less burdensome for them. Prior to engaging in a large RCT, we will first demonstrate that it is feasible to implement an RCT evaluating a time-sensitive intervention that cannot be objectively measured and which is applied in a complex setting.
Keywords: Randomized trial; Research methodology; Research ethics; Mixed methods; Critical care
Consent in critical care: a unique paradigm
The design and implementation of research studies in the Intensive Care Unit (ICU) poses unique challenges including the need to obtain consent for patients who lack decision-making capacity, operationalize protocols under emergency conditions, and study conditions with high associated morbidity and mortality. The majority of critically ill patients are unable to comprehend or communicate at the time they are identified to be eligible to participate in a research study. Consequently, consent for their research participation is typically obtained from SDMs. SDMs may be overwhelmed by the severity of the patient’s circumstances and the ICU environment. Difficulties in obtaining informed consent may reduce opportunities for critically ill patients to participate in research, prolong study implementation [1], limit external validity of study results, and ultimately, delay identification of effective, ineffective, and harmful treatments [2,3]. Consequently, there is a strong need to identify how to best interact with SDMs to make consent encounters more comfortable, credible, informed and less burdensome for them.
Controversial issues in consent procurement in critically ill patients
Debate exists over whether consent should always be required for participation in acute care research and, if required, who should be entrusted to make these decisions [4-6]. The ethical basis for and representativeness of SDM consent for research participation has been questioned amidst evidence suggesting that SDMs make decisions that differ from those patients would have made [7]. Moreover, concern exists as to whether SDMs can comprehend information presented to them with one study demonstrating that 50% of SDMs fail to understand information presented to them pertaining to treatments [8] and another finding that symptoms of anxiety and depression were present in 73% of family members and 84% of spouses [9]. Additionally, family members may not know the patient’s values and preferences [10], and may hold views regarding research participation that differ from those of patients [11]. A systematic review found that SDMs predict patient’s treatment preferences with only 65% accuracy [12]. Another prospective, multicentre study evaluated research participation decisions among critically ill patients, SDMs, and physicians at ICU discharge, and found not only patient-SDM (32% and 42%) discrepancy but also patient-physician (25% and 46%) discrepancy in decision-making in minimal and greater-than-minimal risk studies, respectively [13].
Most patients want their SDMs to be involved in decisions regarding their research participation [13-15]. Whether SDMs experience duress from being actively engaged in the consent process and their desire for autonomy in the decision-making process are important and understudied issues. An Australian study found that parents were highly receptive to being approached for participation in studies, enrolling their critically ill babies in up to 6 clinical studies concurrently [16]. Strategies for SDM decision-making for patient care range from passive, with decisions deferred to physicians, to active, wherein decisions are made autonomously [17-19]. While family members of critically ill patients desire communication with and information directly from physicians, the degree to which SDMs desire physician involvement in research decision-making has not been studied in realtime [20]. A study involving hypothetical scenarios demonstrated that while SDMs were willing to be involved, they were often uncomfortable making consent decisions with over 75% of SDMs wanting to speak to an ICU physician about consent decisions [14]. A Cochrane review of strategies to enhance research recruitment underscored the need for primary research to be conducted on recruitment especially in circumstances where decisions are made by proxies [21].
Approaches to consent procurement in the intensive care unit
In practice, SDMs are approached for research consent in various ways, guided largely by local site norms, protocol-specific directives, legislation, and REB edicts. The Personal Health Information Protection Act (PHIPA) advises that researchers should not make contact with the individual, directly or indirectly, unless custodians (physicians taking care of the patient, nurses or social workers) first obtain the individual’s consent to be contacted [22]. In a prospective study, involving 6 pediatric ICUs and 271 SDM consent encounters, Menon et al. [23] observed higher consent rates when research assistants were introduced by a member of the clinical team prior to approaching the family (89.7 vs. 77.7%; P = 0.04). However, data from a national, prospective observational study involving 23 adults ICUs in Canada found that most SDMs were approached by RCs alone suggesting that introductions of research staff to SDMs may be infeasible in practice [24]. No randomized trial has been conducted to examine how SDMs respond to different approaches for proxy research consent. The lack of evidence to guide best practice underscores the need to systematically study how to best approach SDMs to make consent encounters more comfortable, credible, informed and less burdensome for them.
Study design
A multicentre, pilot RCT comparing different strategies [physician (MD) introduction vs. non-physician (non-MD) introduction] for introducing research personnel and research participation to SDMs (target n=150) of critically ill adults.
Primary objective
To demonstrate the feasibility of an RCT evaluating two methods of approaching (MD vs. non-MD introduction) SDMs for consent for research participation and to conduct a nested qualitative study evaluating SDM’s experience in being approached for consent using the alternative strategies.
Secondary objectives
Among SDMs approached in person or by telephone for consent to participate in a critical care research study by either MD or non-MD introduction, we will:
[1] Describe the proportion of SDMs providing and declining consent. Further, among SDMs in the physician introduction arm, we will compare the proportion providing/declining consent in those provided with a clinical patient update along with an introduction vs. an introduction alone.
[2] Determine the time interval between critically ill patients’ meeting study eligibility criteria and:
(i) SDMs being approached for consent,
(ii) Consent being obtained, or
(iii) Consent being declined,
[3] Elucidate reasons why SDMs provide or decline consent.
[4] Assess agreement between questionnaires completed by SDMs, and the research team (RCs and physicians) probing their acceptance of, and comfort and satisfaction with the approach utilized.
Study population
All critically ill adults admitted to a study ICU who is eligible to participate in any critical care research study in progress at the time of the data collection for which SDM consent is required during regular RC hours will be eligible to participate.
Inclusion criterion
SDMs of critically ill adults, who meet inclusion criteria and have no exclusion criteria to participate in a critical care research study for which in-person or telephone SDM consent (with available contact information) is attempted or required during regular hours will be eligible to participate. When deferred consent and SDM consent are both permitted for study inclusion, we will prioritize procuring consent from SDMs, recognizing that only initial encounters with SDMs will be randomized in the Approach Trial.
Exclusion criteria
1. Since the ability to contact an SDM is inherent in the inclusion criteria, we will exclude circumstances in which no SDM exists, no contact information is available for an SDM, and those circumstances in which contact information exists but is non-functional. Further, we will exclude cases in which we are unable to clarify who the decision maker is.
2. We will limit enrollment to include first encounters to prevent carry over effects. We will exclude any encounters that follow after an initial encounter between the SDM and research personnel has taken place.
3. We will not include SDMs of eligible critically ill adults who die or are transferred out of participating ICUs prior to being screened by a RC.
4. We will exclude SDMs of critically ill patients who are capable of providing first person consent for research participation.
5. We will exclude patients who pre-consent for study inclusion (SDM consent not required)
6. We will exclude studies for which a specific approach for procuring consent has been specified in the study protocol.
7. We will limit studies to those being implemented in the ICU (i.e., not follow up studies for which SDMs are contacted outside of the ICU setting); however, studies operationalized by ICU research personnel in the Emergency Department can be included.
8. We will exclude studies using waived consent
9. We will exclude studies for which consent is not required (e.g., observational studies)
Special consideration: deferred to SDM consent
We are interested in first encounters with SDMs regarding research participation. We will not include circumstances in which patients were enrolled into a research study under deferred consent, and RCs follow-up with SDMs to obtain consent for continued participation (i.e., agreement with a previously made decision is being sought). Similarly, if a patient is becomes eligible for a second research study prior to RCs initially approaching the SDM, we recognize that RCs will want to clarify the ‘deferred consent’ outcome first and consequently we will not randomize these SDMs.
Once eligibility has been determined and an SDM has been identified, RCs will randomize SDM encounters, stratified by ICU, using a central electronic randomization system (rapid.randomization@ gmail.com) with blocked randomization. In the email subject line, RCs will indicate the name of their hospital, ICU and assigned patient identification number. An email response indicating randomization to either (i) an MD introduction or (ii) a non-MD introduction will be returned in 15 minutes or less. If no response is received, RCs will call a pager number and, subsequently, a cell phone number to facilitate randomization. A waiver of consent will be necessary to implement the Approach Trial since it will be infeasible to approach SDMs for consent prior to introducing research personnel to them.
Interventions: approaches for introducing research to substitute decision makers
In the intervention arm physicians will introduce RCs and study participation to SDMs. In the control arm RCs will either introduce themselves or be introduced by a non-physician member of the ICU team. To enhance the likelihood that an MD introduction will occur, we will distribute letters to attending ICU physicians and fellows (specializing in critical care) in participating ICUs informing them that they may be asked to provide introductions and about the content of the introduction. An MD introduction, guided by the letter distributed to physicians or a standardized script for telephone consents, will involve 2 components (1) an introduction of research personnel by name (either in person or by telephone) as a member of the ICU team to the SDM and (2) a statement acknowledging that the patient is eligible to participate in a study that the RC would like to discuss further with them. While both attending physicians and fellows may introduce RCs to SDMs in the intervention arm, we will preferentially request that the most responsible physician (or attending critical care physician) be involved in introductions, where feasible. A study overview is provided in Figure 1.
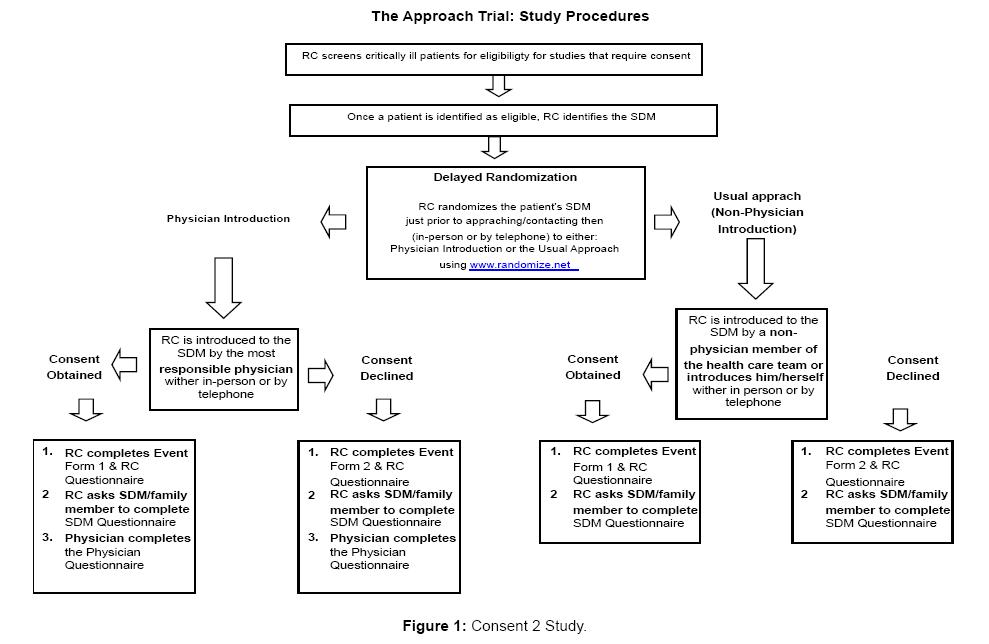
Figure 1: Consent 2 Study.
A two page Event Form (Appendices 1-4) will be completed by RCs when a critically ill patient meets eligibility criteria to participate in a critical care research study that requires SDM consent. Separate Site Forms (Appendix 5) and Study Forms (Appendix 6) will capture information regarding participating ICUs and research studies in operation in the ICUs over the data collection period.
Post encounter questionnaires
After the encounter, RCs will administer 3 to 8 item questionnaires to all individuals involved in the introductions. In the MD introduction arm, questionnaires will be administered to SDMs, RCs, and physicians. In the non-MD introduction arm, questionnaires will be administered to SDMs and RCs. The questionnaires will bear three similar questions probing SDMs, RCs, and physicians comfort and satisfaction with, and acceptance of the approach used to introduce research participation (using Likert Scales from 1-10) [25] (Appendices 7A,8,9). After a consent decision is rendered, we will ask SDMs to complete a second questionnaire probing the main reason for their decision to provide or decline consent and asking about their perceived risk of the study (Appendix 7B). Similarly, we will ask the RC involved in this discussion to specify the main reason in their opinion, based on their interactions with the SDM, why consent was provided or declined.
Pilot study outcomes
Feasibility of the pilot study will be assessed by metrics that reflect our capacity to implement the trial as designed.
Primary outcome
1. We expect that (i) ≤ 15% of physician introductions will be missed due to lack of physician availability (MD introduction arm) and (ii) cross-overs (from either arm to the other) will occur in ≤ 15% on introductions.
Secondary Outcomes
2. Following randomization, we expect that ≤ 20% of introductions will be missed due to inability of the RC to contact existing SDMs once identified (either intervention or control arm).
3. We will obtain at least 70% of initial SDM questionnaires (Appendix 7A) and at least 50% of questionnaires (Appendix 7B) in which consent is explicitly provided or declined.
4. For each initial encounter, we will also record whether consent for the research study was obtained or declined. Failure to return an answer to the RC or time running out (patient is no longer eligible for study inclusion) will be considered to represent declined consents.
Sample size estimation
In a prior study of research recruitment practices, we collected data on 179 consent encounters (140 obtained and 39 declined consents) in 23 ICUs over an approximate 4 week period, or 1-2 encounters per site per week [24]. With 150 encounters, we will be able to assess the potential for crossovers (i.e., physicians being unavailable to facilitate approaches to SDMs in the MD introduction arm or physicians approaching SDMs in the non-MD introduction arm) to adversely impact study implementation across ICUs.
We will use descriptive statistics, including measures of central tendency and proportions for continuous and binary variables respectively, to summarize results. We will report time intervals using mean and standard deviation (alternatively, median and interquartile ranges depending on the distribution of the data).
Primary analysis
We will tabulate the proportion of encounters wherein (i) a physician was not available and (ii) a strategy other than the assigned strategy was utilized.
Secondary analysis
We will compute the proportion of introductions (after randomization) that were missed due to inability to contact an identified SDM and the proportion of returned SDM questionnaires (initial and subsequent). We will compare the proportion of consents (i) obtained and declined in both arms and (ii) with and without a patient update accompanying a physician introduction in the physician introduction arm using the Chi-square test (or Fisher’s Exact test for expected values <5). We will summarize reasons why SDMs provided or declined consent. We will compare binary and continuous measures between consent obtained and declined outcomes using the Chi-square test (or Fisher’s Exact test for expected values <5) and Student’s t-test, respectively. We will assess acceptance of and comfort and agreement with the approach utilized to introduce RC and the notion of research participation to SDMs by comparing responses of RCs and SDMs to post encounter questionnaires using the Mann Whitney-U test. Similarly, we will evaluate participant’s (RCs, SDMs and physicians) acceptance of and comfort and agreement with the approach utilized by comparing their responses to post encounter questionnaires using the Kruskal-Wallis test. Finally, we will compare the proportion of responses between SDMs and RCs as to the rationale as to why consent was obtained or declined using the Chi-Square test (alternatively, Fisher’s Exact test). All analyses will be conducted using SAS 9.1 (SAS Institute, Gary, North Carolina).
Research question
“What is the experience of SDMs in being approached for consent for research participation?” To address this question, we will conduct 12 one-on-one semi-structured interviews with SDMs (6 MD and 6 non- MD introductions) to explore the beliefs, values, and perceptions that provide the context for their experience.
Inclusion criteria
We will include SDMs approached for consent in the Approach Trial with a consent outcome (obtained, declined) and who agree to participate in the qualitative study.
Exclusion criteria
The exclusion criteria for the qualitative study will be identical to the quantitative study. In addition, we will exclude SDMs who both provide and decline consent for research participation during the same encounter and those randomized to one approach who cross-over to the alternate approach as the context of the encounter may be different. We will also exclude SDMs who were approached on a subsequent occasion (e.g., second or third approach) prior to the interview being conducted.
Methods: qualitative approach trial
A trained interviewer (student) will use a structured interview guide to conduct in-depth SDM interviews at 2 centres [Mount Sinai Hospital and St Michael’s Hospital] using modified grounded theory. Interviews will be conducted as close to the encounter as is agreeable to SDMs given the need for the initial approach to be the topic of discussion. The interviewer will ask SDMs to place themselves back to the time when they were first approached about research. SDMs will provide written consent just before the scheduled interview. A priori, we anticipate that it may be more challenging to consent SDMs for participation in the qualitative study that declined consent for a patient to participate in a research study.
Interviews
We designed separate interview guides to reflect the alternative approaches (MD vs. non-MD approach) being evaluated. We will include both general and open-ended questions in the interview guides to enable SDMs to describe the consent event and recall specific milestones and aspects of the consent process. Questions in the two interview guides will be similar; however, each guide will have one unique question pertaining to physician involvement. We will pilot test the interview guides for clarity and comprehension with 2 RCs and 2 SDMs before administering the interview guide. Interviews will be approximately 1-hour in duration and audio-recorded. Taped interviews will be transcribed verbatim with identifiable information removed by the transcriber. Transcribed records will be entered manually and/or in NVivo™ for analysis.
Coding and analysis: qualitative approach trial
All data will be coded by two coders. Coding of each line manually and/or with NVivo™ software will facilitate identification of initial concepts. During coding, emerging codes will be identified. Selective and axial coding will be used to delineate codes that reappear frequently and to sort data. We will utilize codes to identify categories which will shape development of the analytic framework (emerging themes) until theoretical saturation is achieved. Thematic analysis will be conducted upon completion of all interviews to identify emerging themes that will reflect the factors, processes and conditions during the consent encounter of relevance to SDMs when approached.
Approaching SDMs for proxy consent is qualitatively different than directly approaching patients for consent for research participation While PHIPA legislation provides guidance on how to approach consent providers [22], its recommendations are not evidence-based. Moreover, the legislation does not specifically address proxy decisionmakers. Several pragmatic issues introduced by the need to interact with SDMs in the acute care setting add additional layers of complexity to ICU research implementation. While SDMs are approached daily for proxy consent in the ICU, little is known about the optimal manner to introduce research to them and whether physician involvement enhances their experience. Consequently, there is a strong need among stakeholders in clinical research to better understand how to best interact with SDMs for proxy research consent.
The Approach Trial is novel. First, it is the first randomized trial designed to compare alternative strategies for introducing research to SDMs of critically ill patients and directly addresses an appeal to conduct primary research on strategies to enhance recruitment in areas involving proxy decision-makers [18]. Second, unlike most critical care trials the focus of this RCT and the unit of randomization will be the SDM. Third, this trial will evaluate real-life experiences of SDMs, RCs and physicians. Fourth, as a mixed methods trial, this study includes a quantitative component to evaluate the feasibility of implementing a trial comparing two strategies for introducing research to SDMs and a qualitative component to understand SDM’s experiences in being approached for proxy consent.
In the Approach Trial, we aim to evaluate whether centres can implement the trial as designed. Conduct of a trial comparing two different strategies for introducing research to proxy decision-makers poses several unique challenges. First, research personnel must randomize SDMs prior to approaching them. This aspect of protocol implementation imposes a change in RC’s practice as they typically introduce themselves to SDMs as opportunities arise, clarify their status as decision-maker and then randomize patients (as opposed to SDMs). Second, RCs will need to locate a physician to make an introduction in the MD introduction arm. ICU physicians are often engaged in admitting, evaluating or stabilizing critically ill patients, performing procedures, educating trainees, or meeting with families. Reduced physician availability may constrain RC’s time and their ability to coordinate introductions with SDMs. Opportunities to interact with SDMs in the ICU may be transient and limited by the need to provide nursing care, perform procedures, or transport patients for investigations or interventions. The introduction strategies may complicate normal research processes and impact RC workload. To enhance the likelihood of an MD introduction occurring, we will distribute letters to attending physicians and fellows informing them that the trial is in progress, advising them that they may be asked to make an introduction, and highlighting the components of an MD introduction. Third, to compare participant’s comfort and satisfaction with, and acceptance of the alternative approaches, we will create three questionnaires with identical Likert scales [25]. The questionnaires will be administered to SDMs, RCs and physicians (MD introduction arm) and SDMs and RCs (non-MD introduction arm). Fourth, unlike a medication or a device, the interventions must be perceived by SDMs as they are intended to be delivered and cannot be objectively measured. To address this concern, we plan to conduct a nested qualitative study of SDMs experiences in being approached. Five, the practicalities of conducting a trial on research processes in the ICU, while novel, are unknown as no randomized trial has previously been conducted.
Strengths of the Approach Trial design include use of central randomization, allocation concealment, and conduct of an intent-totreat analysis to enhance internal validity. We will include consecutive SDMs of eligible critically ill patients who require SDM consent for research participation. Inclusion of consecutive SDMs will enable us to quantify protocol violations and cross over rates, identify barriers to approaching SDMs after randomization, and the frequency with which SDMs are randomized, approached but never return a response. To prevent carry-over effects, we will limit introductions to first encounters. To standardize MD introductions, we will include a script for both telephone introductions and a letter to guide in-person introductions. To understand how participants involved in encounters perceive them, we will administer questionnaires to all involved participants. To gain insight into the factors, processes, and conditions of importance to SDMs, we will conduct a fully nested qualitative study within the Approach Trial.