Journal of Theoretical & Computational Science
Open Access
ISSN: 2376-130X
ISSN: 2376-130X
Research Article - (2017) Volume 4, Issue 2
Models of gene regulatory networks (GRN) have proven useful for understanding many aspects of the highly complex behavior of biological control networks. Randomly generated non-Boolean networks were used in experimental simulations to generate data on dynamic phenotypes as a function of several genotypic parameters. We hypothesized that the topological component of network genotype could be an obstacle to the discovery of mathematical formulas that can predict certain phenotypic parameters. Our data support that hypothesis. We quantitated the effect of topological genotype (TGE) and determined its influence on a number of dynamical phenotypes in simple and complex multi-gene networks. For situations where the TGE was low, it was possible to infer formulas to predict some phenotypes with good accuracy based on number of network genes, interaction density, and initial conditions. In addition to formulation of these mathematical relationships, we found a number of dynamic properties, including complex oscillation behaviors, that were largely dependent on genotype topology, and for which no such formulas were determinable. For integrated measures of gene expression state, we observed a variety of oscillation patterns, including stable, periodic cycling with a wide variety of period length, aperiodic cycling, and apparent chaotic dynamics. It remains to be determined if these results are applicable to biological gene regulatory networks.
<Keywords: Gene regulatory network, Biological, Mathematical analysis, Gene expression
Gene regulatory network (GRN) architecture is believed to play a crucial role in biological function [1,2] and innovative evolutionary transitions [3]. Research into the relationship between innovation (necessary for evolution) and network robustness has found important clues to the mechanisms of evolution in both biological GRNs [4-8] and model GRNs [9-14].
Our goal has been to explore the existence of fundamental principles that govern the behavior of these complex regulatory networks irrespective of their relationship to evolvability or robustness. Mathematical analysis of random Boolean networks and other models has provided some insights into the expected dynamical behavior of networks in ordered, periodic, and chaotic regions [15-19] based on network genotype and interactive rules.
Our approach uses non-Boolean, iterative model network systems as generators of experimental data [20-22]. This kind of approach has been used to define critical modular topologies within networks [23- 25], for the theoretical determination of dynamical network properties [13,26,27], as well as to investigate the evolution of evolvability [28] and other properties of complex networks.
Given the highly complex nature of the behavior of even very simple models of a few genes, we were interested in the basic dynamical properties of model networks as a function of several parameters of network complexity, including the total density of gene-gene interactions [29,30], as well as genotypic topology.
The basic assumption guiding this work is that it is possible to formulate deterministic laws that govern the behavior of complex model networks. We hypothesize that the ability to make quantitative predictions concerning phenotypic endpoints of network activity is an inverse function of the degree to which network topological genotype plays a role in network dynamics. The likelihood that this hypothesis is true has been shown in previous work [23,31-33] that demonstrated the importance of specific gene-regulatory mapping in different networks with identical quantitative genotypic parameters (e.g., identical interactive densities). However, the precise role of genotype topology in the predictability of dynamical network phenotypic endpoints has not been investigated in depth in a system of model networks.
In order to explore the above hypothesis, we constructed model networks in two categories of interactive complexity. Gene interactions within a network can be either activating or suppressive. The resulting complexity of phenotype, defined as the specific dynamics and degree of gene expression in the networks, varies considerably depending on whether both or only one type of interaction is present in the network genotype. We found that networks that include only gene activation by other genes (“Activation networks”) are simpler and useful for investigation of potential quantitative laws governing phenotypic output dynamical behavior. Networks that include both activation and suppression of gene expression by other network genes (“Compound networks”) are far more complex, but also more relevant to biological gene regulatory networks.
We investigated the dynamics of both network types by allowing each gene in the network to assume an additive, continuous state of expression (as opposed to the “on or off” states of the Boolean approach). We analyzed gene state variables (defined in Methods) corresponding to an integrated view of the state of all the genes in the network as a function of time, given all possible initial conditions. Our goal was to find reproducible, predictive patterns of gene expression related to the quantitative aspects of network architecture such as number of genes, interaction density, and initial conditions. We were able to explore the relative effects of topological genotype vs. measurable quantitative network parameters on these state variables.
The phenotype of each network at time t is a function of the expression state Si,t of each gene i in the network. The equilibrium phenotype Si,eq is either the expression state of each gene when the gene trajectory (see Methods) reaches stability, or, for oscillating trajectories, the average value of Si over one cycle.
In our approach, the state of each gene at any time point (Si,t) is represented by the sum of all interactions from other network genes that act upon it. This value will be an integer and may be negative. Each gene can contribute a positive (activation) value of +1, a negative (suppression) value of -1, or a no-interaction value of 0 to the state of each of the other genes in the network. Genes only exert their effects on their target genes if their own expression state at time t is greater than 0. The distribution of these interactive values for each gene with every other gene is defined as the network genotype, as illustrated by the example shown in Figure 1.

Figure 1: 5-gene model regulatory network. A) Matrix array showing numerical values for each interaction between all genes. The effect of genes in the rows on genes in the columns is given by 0 (no interaction), +1, or -1. Self-regulation is not included. B) A diagram of the interactive network shown in A, with green arrows showing activating interactions, and red blunted arrows showing suppressive interactions. Green double arrows (between Genes 1 and 5 and between Genes 4 and 5) indicate reciprocal activation, and bicolor arrows (between genes 3 and 4, Genes 2 and 3, and Genes 2 and 5) indicate inverse reciprocal interaction, where one gene activates another gene that suppresses it. For example, Gene 2 suppresses Gene 3, activates Genes 1 and 5, is activated by Gene 3, and suppressed by Gene 5.
We followed the previous approach of Wagner and his colleagues [34-36] by constructing random model regulatory networks in the form of an NxN square matrix (wij) of N genes. The total number of potential interactions in this type of network is N2. Excluding autoregulation [25,37,38], the diagonal is fixed at 0, giving the total number of interactions (and cells in the model matrix) as N2 – N.
Model networks, like biological regulatory circuits, operate dynamically, with feedback loops and complex interactions taking place over time. This is simulated in model network analysis by running iterations of the calculation of network states given the changing expression state of each gene as a function of time. Stable or periodic patterns for Si,t emerge from time-dependent feedback mechanisms of genes acting on each other.
Genotypes
Each network’s genotype is defined by the number or density of activating and suppressive interactions as well as the specific topology of the network matrix wij. Since there are 3 possible interaction variables (0, +1, and -1), the total number of possible networks for N genes is 3N2 −N . As N grows, this number becomes very large; for 5 genes, there are more than 3 billion possible network genotypes. The example in Figure 1 shows a network of 5 genes with 9 activating and 5 suppressing interactions.
Density parameters
We define some density parameters that give an approximation of the degree of complexity of the network in terms of the number of interacting genes. If A is the number of genes that activate other genes, and U is the number of genes that suppress other genes, then 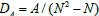 is the density of activating genes, and
is the density of activating genes, and  is the density of suppressor genes. The total density of interacting genes in a network, DT, is the sum of DA and DU.
is the density of suppressor genes. The total density of interacting genes in a network, DT, is the sum of DA and DU.
For analysis of Compound network outputs, numbers of both activators and suppressors need to be considered. Net density, a function of the difference between the number of activating and suppressing interactions, is a useful measure for these networks. Net density (Dn) is defined as:
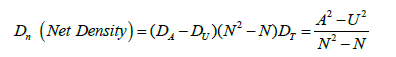 (1)
(1)
Genotype components
In practice, a description of the genotype (such as the matrix shown in Figure 1) falls into two categories: quantitative parameters and topological mapping. The genotypic quantitative parameters include the total number of genes (N), the density of interactive elements (DA, DU, DT, and Dn), and the number of genes expressed in the initial condition of the network (G0). Each of these parameters is easily defined. In contrast, topological mapping of the network, which defines the precise set of rules governing gene/gene interactions, requires the use of a lookup table, as is done for simple Boolean networks. In small networks involving three genes, it is possible to assign interactions to one or more “modules” (e.g., positive feedback loops), and quantitative assessments of the effects of topological genotype are possible [13,39]. This is extremely difficult when N=4, and not possible for N=5 or higher.
Initial conditions
Any given network will produce different phenotypes depending on the initial condition of the network at t=0. The initial network condition depends on the influence of factors external to the network, such as specific transcription factors that can target one or more network genes. In order to represent each of the 2N possible combinations of initially expressed genes, we use a binary code system, where 1 indicates initial expression and 0 indicates initial non-expression of each gene. For a 5-gene network, E (the initial gene expression vector) can be from 00000 to 11111. Thus when E=01101, Genes 2, 3, and 5 are initially expressed, and genes 1 and 4 are not expressed at t=0. The number of genes that are expressed in the initial condition is given by G0, which is defined as:
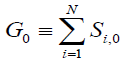 (2)
(2)
Thus, for E=01101, G0=3. For each value of G0 (from 1 to N) we can use the combinatorial formula to find that there are G0CN values of E. For example, for a 5-gene network, 10 of the possible 32 initial conditions will have G0=3.
Phenotypes
As in all biological systems, the phenotype of any network is the sum of all the characteristics of the system, which derive from the state of each gene at any given time point. Any network phenotype can be represented by the time-dependent and initial condition-dependent state of each gene in the network. For some purposes, such as studies of robustness, evolvability, or the effects of mutational perturbations on phenotypes, this is an appropriate and useful measure of phenotype. It is more difficult to analyze dynamical behavior of more than one gene at a time in order to characterize the influence of genotype on the phenotype of the overall network. We therefore use two integrated measures of network phenotype that take into account the state of all genes at equilibrium.
We define HG0 as the average of all gene state values, Si,eq, for each of the N genes at a particular value of G0.
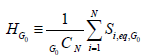 (3)
(3)
For further integration of network dynamics, we define W as a single number that represents the sum of HG0 values over all possible 2N initial conditions (or all values of G0) of the network.
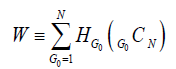 (4)
(4)
For oscillating Compound networks, other phenotypes, such as the period and amplitude of oscillation and the proportion of gene trajectories that show stable or oscillating behavior were also determined. Gene trajectories are defined as the time course of the expression state of each gene as a function of E. The number of gene trajectories for any network is equal to N2N.
Network construction and analysis
All model networks were produced using a random number generator to assign each cell in the network matrix a value of 0, 1, or -1. Networks were then analyzed using computations with a MATLAB (version 8.6) [40] program to produce S for each gene as a function of time and E. The value of Si,t was calculated as the sum of the effects of all other genes in the network that affect gene i. This allows the final state of each gene at any time point to vary from negative to positive integers. An iterative algorithm was used to model time variables, with each iterated value of S at iteration t dependent on the value of S at t-1 [34]. If Si,t-1 is>0 (expressed) it will exert its genotypically determined effect (activation or suppression) on its target genes, j. If Si,t ≤ 0 (not expressed), gene i will have no effect on the other j genes.
Values of HG0 and W (defined above) are also calculated by the program as a function of time. Calculations were done using all 2N values of E.
Topological genotype effect
We used analysis of variants (ANOVA) to quantify the degree of importance of genotype vs. other input parameters. When comparing a given output (dependent variable) between networks with identical quantifiable parameters, the ratio of within-group variation to the total variation (as given by sum of squares in ANOVA) gives a good approximation of the influence of topological genotype on the output.
Role of genotype topology
To test our hypothesis that prediction of network behavior is possible only to the extent that the phenotype in question is largely independent of topological genotype, we began the analysis of dynamical network behavior with the simpler Activation networks. First we needed to determine to what extent each measured phenotypic output is dependent on topological genotype vs. quantifiable genotypic inputs.
As shown in Table 1, the values of Topological Genotype Effect (TGE) vary from close to 0 to 80%, depending on the kind of network (Activation vs. Compound) and the output parameter being analyzed. We observed an inverse correlation between the TGE and the correlation coefficients (R2) for curves of output vs. independent input variable (data not shown). This confirms our assumption that for measures with a high degree of topological genotype dependence, the usefulness of quantitative models to predict outcomes will be approximate at best.
| Phenotype Parameter | Activation | Activation |
|---|---|---|
| Si,t | 0.46 | 0.72 |
| HG0 | 0.015 | 0.24 |
| W | 0.026 | 0.18 |
| Period | - | 0.80 |
| Amplitude | 0.64 | - |
| %Stable | 0.56 | - |
Table 1: Values of TGE for Various Phenotypes.

Figure 14: No relationship between the fraction of stable gene trajectories and net density. This was also seen for all other measures of interaction density.
A more detailed examination of the data shows that for the determination of HG0, there are some trends in the degree of the TGE for both Activation and Compound networks beyond the overall strength of the effects seen in Table 1.
For Activation networks, Figure 2 indicates that there is a trend toward lower TGE as the number of genes expressed at the initial condition (G0) increases, so that at the maximum (G0=N), TGE is 0. This implies that any predictive formula for an output will be more accurate when more genes are initially expressed, which is in fact what was observed. For the maximum case, when G0=N (and TGE=0), we found that experimental values of HG0 were always perfectly predictable to be equal to A+G0.

Figure 2: Influence of the number of genes expressed in the initial condition (G0) on the Topological Genotype Effect. Data shown is for calculation of ANOVA for 20 Activation networks with DA=0.1 to 0.25.
We determined the influence of increasing density of activators and suppressors on the Topological Genotype Effect in Compound networks by analyzing the TGE in replicate networks with one or the other interaction density held constant. We found a strong positive association between TGE and density of suppression interactions, while the density of activating interactions had no effect on TGE (Figure 3). The observation that having more suppressors in a network increases the topological effect may not be surprising when the strong effect of the topology of suppression on phenotype is considered; but the lack of any effect of activator density is not easily explained.

Figure 3: The influence of activating and suppressing interactions within networks on the Topological Genotype Effect on HG0. Blue triangles are activating and black circles are suppressive interactions; the line is the least squares fit for suppressive interactions (R2=0.85). Each point represents the TGE when one of the interactive elements was held constant (at the value shown on the x axis) with varying numbers of the other element.
Gene expression state as a function of activation density
The data for TGE values in Table 1 suggest that values of Si,t for each gene are strongly dependent on topological genotype (more so for Compound than for Activation networks), and should not be accurately predictable by any quantitative model if our hypothesis is correct. On the other hand, based on these data showing a less than 3% contribution of the topological genotype to the determination of the integrated gene expression parameters HG0 and W in Activation networks, we would expect to be able to formulate quantitative relationships for the behavior of these phenotypic outputs using quantitative genotypic parameters. While the TGE contribution for HG0 and W is higher in Compound networks, it might be possible to develop approximate models for these as well. Based on the strong TGE shown in Table 1 for oscillating period, amplitude, and percent of stable gene trajectories, we would not expect to find any useful quantitative relationships for these phenotype outputs.
Single gene expression states
Figure 4 illustrates the relationship between Seq for a single gene (at a single value of G0) and activation density. For both Activation and Compound networks, the expected positive relationship is seen (increasing the density of activation interactions would logically increase the overall expression levels of most genes), but the scatter of the data makes it clear that accurate predictions of the precise level of expression are not possible. As expected, this is even more true for Compound networks.

Figure 4: Seq for one gene with G0=4 as a function of activation density. A) Activation networks. As expected, a trend is seen for increased expression with increased activation density, but variability (due to the TGE) is high. The regression line shown has R2=0.66. B) Compound networks. Compared to A, there is more variation, with R2=0.3, although a trend of increasing Seq (averaged over one cycle period for oscillatory gene trajectories) with increasing net density of activation is still observed.
Integrated network expression states
To examine the relationships between integrated measures of network expression HG0 and W and quantitative genotype parameters of activation density and G0, we analyzed 310 Activation networks comprised of 4, 5, and 6 genes, as well as 480 5-gene Compound networks. As expected, we observed strong correlations between both integrated phenotypes and these genotype parameters in Activation networks.
We found that the derived quadratic formula shown in equation 5 fits the actual experimental data better than any strictly empirical model, over the whole range of DA and G0 values for several values of N. Equation 5 allows us to accurately predict within a 5% margin of error the value of HG0 for any network at each value of G0, given the total number of genes (N) and the density of activators (DA) in the network.
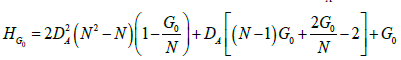 (5)
(5)
This formula was derived from basic principles of network dynamics. The maximum value of HG0 for any Activation network is A+G0, and the minimum value is A(G0/N). We found that the experimental values of HG0 were dependent on the probability of multiple interactions between the genes in the network. For example, if two genes activate each other, the value of HG0 will be higher than if each activation is independent of other genes. The probability of such interactions rises to 1 when G0=N, and also rises for all values of G0 as DA increases. We cannot derive an exact determination of this probability for a general network, since the number of potential interactions in N=4 and higher networks is too large for precise analysis. We used an approximation for the multiple interaction probability, and developed a formula whereby HG0 is the weighted average of the maximum and minimum values as a function of DA, N and G0. The weights are determined by the approximate probability, which is also a function of DA and N. The resulting quadratic relationship between DA and HG0 fits the data as illustrated in the examples of Figure 5 for all values of N, DA, and G0.

Figure 5: Illustration of close agreement between experimental values of HG0 (points) and theoretical predictions based on equation 5 (lines). A) N=4; blue triangles - G0=2, black circles - G0=4. B) N=5; blue triangles - G0=1, black circles - G0=4. C) N=6; blue triangles - G0=2, black circles - G0=5. Blue dashed lines and solid black lines are the corresponding calculated values of HG0 using eq. 5.
Given that W is defined as the sum of HG0 over all values of G0, we were able to derive a formula for W as a function of DA and N that is also highly predictive of the experimental value of W for all Activation networks. The derived formula for W (equation 6) fits the experimental data very well, as shown in Figure 6.

Figure 6: Experimental confirmation of the predictive utility of eq. 6 for values of W for Activation networks. Points represent experimental data, and the line is derived from values calculated according to eq. 6.
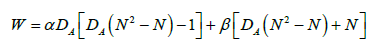
where
α=2N-2
β=2N-1
Compound networks
For Compound networks, time course dynamics are more complex and do not show any simple relationship with total interaction density (DT). Since values for HG0 and W are expected to rise with increasing number of activators and with decreasing number of suppressors, it is a reasonable hypothesis to test whether the difference between the activator and suppressor densities is correlated with W and/or HG0. However, since this difference alone does not take into account the total number of either activators or suppressors, it isn’t surprising that no correlation was seen with the difference DA - DU. We corrected for this by calculating the net density (Dn) metric, which is the difference between the number of activators and the number of suppressors times the total density (DT) (see Methods). As shown in Figure 7, plots of Dn vs. HG0 and W do show increasing trends.

Figure 7: Fit of experimental data (points and error bars) to the theoretical values obtained from eq. 7 in 84 Compound networks. Open circles and solid line are for G0=1, and solid blue points and dashed line are for G0=4.
For Compound networks, we found an empirical formula (equation 7) based on analysis of 84 networks that relates DA, DU, and G0 to the average value of HG0 over all time points.
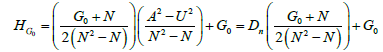 (7)
(7)
As seen in Figure 7, the theoretical results from equation 7 give only an approximate fit to the experimental data, with a median error rate of 24%. We were also able to formulate an empirical relationship between the integrated single state value of W for Compound networks and the quantitative parameters of N and interaction net density (Dn) as follows:
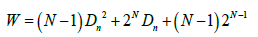 (8)
(8)
As shown in Figure 8, this empirical relationship gave an approximation for the experimental values of W over a wide range of Dn with an error rate of about 60%. Comparison of Figures 5 and 6 with Figures 7 and 8 confirms that the degree of variation of HG0 and W is much higher for Compound networks than for Activation networks. It should be noted that the formulas in equations 7 and 8 for Compound networks do not reduce to the equations for Activation networks with DU=0. In fact, it was not possible to find any formula that would fit both Activation networks using DU=0 and Compound networks. This strongly suggests that there is a qualitative difference in the way these two network types operate: Activation networks do not behave simply as Compound networks with DU=0 for computational purposes.

Figure 8: Experimental values of W as a function of Dn (blue circles) with the theoretical values from eq. 8 (black line).
Oscillation dynamics
For Activation networks (DU=0), we found that all genes reach a fixed steady state within a few iterations and Si,t becomes a constant=Si,eq. All Activation networks are examples of “viable networks” [35] that do not exhibit oscillations in state values at any level of analysis.
Oscillation period
For Compound networks, dynamical patterns are far more complex. The behavior of single gene states was highly dependent on exactly which genes are initially expressed, meaning not only on values of G0, but on specific values of E. For example, Figure 9 shows the dynamics for 5 genes in the same network (the one pictured in Figure 1) for two values of E with G0=2. In Figure 9A (E=00101), each of the genes oscillates at first, and then they all reach a stable state. The same network with the same value of G0=2 but with E=10001 produces a very different pattern for these genes, as shown in Figure 9B.

Figure 9: Dynamic time course behavior for a Compound network with G0=2. A) E=00101, five genes shown; all undergo early oscillations and then settle into stable behavior. B) The same network with E=10001, with dynamics of four of the five genes shown. Only one gene reaches a stable expression state; the other three oscillate continuously.
The TGE for the dynamical behavior of Compound networks is quite high (Table 1), and therefore, if our hypothesis is correct, we would not expect to be able to formulate precise mathematical relationships between any of the oscillatory phenotypic outputs based on quantitative network parameters. After running 480 Compound networks, we found that the distribution of the length of oscillating periods (Table 2) was far from linear. Between one fifth and one quarter of Compound networks actually showed stable, non-oscillating dynamic values of W. For 19 (4%) of the networks, we found evidence of aperiodicity. Four of these aperiodic networks passed the 0-1 test for chaos [41,42] with 10 replicate runs giving R values>0.99. The longest repeating period we observed was 210 iterations. Other rare periodic oscillations present in less than 1% of the networks tested included period lengths of 3, 42, 120, 140, 7, 10, 30, 5, 14, 24, 28, and 180 iterations.
| Period Length | No. | % |
|---|---|---|
| 0 | 108 | 22.5 |
| 4 | 74 | 15.4 |
| 2 | 67 | 14.0 |
| 60 | 61 | 12.7 |
| 20 | 52 | 10.8 |
| 12 | 48 | 10.0 |
| 6 | 21 | 4.4 |
| Aperiodic | 19 | 4.0 |
| 84 | 5 | 1.0 |
Table 2: Distribution of Oscillating Period Lengths for W in 480 Compound Networks.
Figure 10 shows three examples of the periodicity of these Compound networks. As shown in Figure 11A, there is no discernible relationship between average period length and net density (Dn). However, we did find a surprising relationship between the average period length and total density DT (determined by the total number of interactions, both activating and suppressing (Figure 11B). The same pattern was also seen when individual values of periods were plotted against DT (Figure 11C), but not when plotted against Dn (not shown). No other phenotypes showed any relationship with DT.

Figure 10: The complex oscillatory patterns of three Compound networks. A) Oscillatory period=42 iterations. B) An example of a commonly seen period of 60 iterations (present in about 13% of Compound networks). C) An aperiodic network, with an appearance of chaotic dynamics. This network gave a consistent R value>0.99 when subjected 10 times to the 0-1 test for chaos [41,42].

Figure 11: The relationship between oscillation period length and interactive densities in Compound networks. A) points are average period length at each value of Dn for 480 Compound networks. B) Points are the same as in A, but the density measure is DT, the total density of activators and suppressors. C) Points are the actual values of periods of individual networks (not averaged). Each point represents many networks. The eight points at the top (at Period=330) represent 19 aperiodic networks, whose minimal possible period length is greater than 330 iterations. In both B and C there is a demarcation at a value of DT=approximately 0.4, below which no networks show long periods or aperiodic behavior.
Figures 11B and 11C indicate that for networks with total density higher than about 0.4, the general pattern of period lengths is random, and varies from 0 to aperiodic (shown on the chart as>330). However, for the lower density range of DT below 0.4, there is a clear exponential trend for average lengths of network periods, and values do not exceed a period of 20 iterations (Figure 12). Oscillating network period was the only phenotype parameter in Compound networks that showed a relationship with total interactive density, regardless of the type of interaction.

Figure 12: An exponential relationship between the average period length and DT at values of DT below 0.4 for 28 Compound networks. The curve is the best fit (least squares fit) of the data using an exponential relationship.
Oscillation amplitude
Unlike all other phenotypic parameters, the amplitude of oscillations for W for Compound networks shows a decreasing trend with increasing net density, suggesting a direct relationship of amplitude with gene suppression. As expected from the strong topological genotype effect shown in Table 1, this trend is not precise enough to allow for quantitative formulations, and there is a great deal of variation, as shown in Figure 13. It is apparent from the figure that all such networks with amplitude=0 (in other words, non-oscillatory, stable networks) Dn is ≥ 0, confirming that activation interactions are more closely correlated with stability than are suppressive interactions.

Figure 13: An inverse correlation between oscillation amplitude and net density. The line is the best fit linear regression line.
Proportion of stable trajectories
For Compound networks, some fraction of the N2N gene trajectories may be oscillating, and others will be stable. Since values of HG0 and W will oscillate if even one such gene trajectory oscillates, networks that exhibit stable values of HG0 or W have 100% stable gene trajectories. The proportion of stable to oscillating trajectories is not predictable from density and other quantitative genotype values. The Topological Genotype Effect for this phenotype is 0.56 (Table 1). We found no trends for the fraction of stable gene trajectories as a function of any density measure (Figure 14). The figure shows that while some Compound networks have all of their genes stable at all initial conditions, indicated by the fraction of stable gene trajectories=1.0 (the same networks with amplitudes=0), there are no networks with mostly oscillatory gene trajectories. In fact, all networks have at least 55% of their gene trajectories showing stability under all initial conditions. Thus, even for Compound networks with erratic aperiodic dynamics in values for HG0 and W, the majority of the individual gene state trajectory dynamics are stable.
The overall purpose of the investigation reported here was to determine whether, and under what circumstances, it might be possible to formulate mathematical models to fit the behavior of complex regulatory networks. Our strategy was to approach this goal by initially examining simpler networks (Activation networks), which lack any suppressive interactions between genes within the network. This proved to be a successful approach, since it allowed us to find several features of interacting regulatory networks that are important in any kind of theoretical modeling. For example, it became clear that precise prediction of the dynamical behavior of individual genes was not possible without taking into account the precise topology of the network genotype.
The importance of specific topology in determining phenotype outcome in GRNs has been observed by other investigators [31,43], although it has also been found that topology alone is not sufficient to make accurate evolutionary predictions [32]. This is consistent with results of Payne and Wagner [33], who proposed that form and function in GRNs appear to have no consistent relationship.
We confirmed the hypothesis that genotype topology plays a critical role in network phenotype determination by correlating the strength of the influence of the topological character of networks with the precision of quantitative formulas for prediction of phenotypic outcomes. To do this, we used the metric of the strength of the Topological Genotype Effect (TGE) in determining each of the possible phenotypes.
We observed an inverse relationship between the TGE and our ability to find predictive equations for several phenotypes of networks. For stable Activation networks with no suppressor interactions, HG0 and W are quantitative representations of the final fixed phenotype. The theoretically derived equations for these integrative measures of network gene expression as functions of quantitative genotype parameters were accurate to within less than 5% error of experimental results for Activation network simulation runs. Gene number, activator density, and number of genes expressed at initial conditions were the quantitative input parameters used in these equations (5 and 6).
As expected, the value of TGE for all phenotypes was higher for Compound networks than for Activation networks. While empirical equations were moderately accurate for average outputs of HG0 and W, the phenotypic variability of Compound networks with different topologies prevented the prediction of outcomes for specific networks. The extent of the TGE in Activation networks proved to be a function of G0, meaning that the more genes are expressed at the initial condition, the less effect topology has. This seems logical, since if only one gene is initially activated, its precise position in the matrix is of great importance for gene expression trajectories. On the other extreme, when all genes are initially expressed, their positions in the matrix are of no relevance.
Some investigators treat dynamically unstable model networks as non-viable [14,29,38], but we chose to retain such oscillating (cycling) models in our analysis because of their relevance to biological GRNs, and because of their interesting dynamical behavior as a function of time.
Oscillating values of the state of expression of individual genes (Si,t) are common features of model GRNs [44], and have also been found in some biological GRNs [5]. Like others [44,45], we found that a majority of Compound networks, which include suppressive interactions, show oscillation of integrative gene expression metrics (HG0 and W) for most initial conditions. Consistent with results for Activation networks, we found a general trend for higher levels of expression with increasing ratio of activation to suppressive interactions.
We found a surprising differential effect of activation vs. suppressive interactions on the TGE in Compound networks. The density of activators had no influence on the strength of TGE, with a constant value of about 0.25 for all activator densities from DA=0.05 to 0.45. On the other hand, increasing the density of suppressors over the same range increased the TGE in a linear manner, with a value of 0.06 at DU=0.05 to 0.55 at DU=0.45.
The relationship of transcriptional repressors to oscillating networks has been previously studied [45,46]. Negative feedback loops (analogous to gene expression suppression) have been associated with oscillating behavior in many biological systems [47], including E. coli [48], the Neurospora circadian oscillator [49], the cell cycle [50], the lac operon system [51] the p53-mdm2 system [52] and NF-kappa B translocation [53]. Boolean approaches have also been used to model circadian oscillations [54], which are the most studied example of biological periodic oscillations.
Our results show a more complex relationship between gene expression suppression and oscillatory behavior in these model gene regulatory networks. There was a weak trend to higher amplitudes for oscillation of W with increasing suppression density, which is difficult to explain. Contrary to expectation, we saw no relationship at all between any measure of interactive density and the proportion of individual gene trajectories exhibiting oscillatory or stable dynamics. We also noted that the majority of gene trajectories in all networks, even those exhibiting very complex behavior at the integrated level of W, were actually stable, and that many networks with long oscillation periods for HG0 and W included only a small proportion of cycling individual gene trajectories over all initial conditions. This result suggests that in gene regulatory networks, oscillatory behavior of a relatively small number of genes can have a cascading or amplified effect on the network as a whole.
Suzuki et al. [39] found a relationship between the relative density of suppressing to activating interactions and chaotic dynamics in small gene regulatory circuits. Our results confirm these observations in that most of our networks with suppressing interactions showed oscillatory (unstable) behavior for W. However, the relationship of the complexity of dynamics to the density parameters of the networks was highly dependent on topological genotype (TGE=0.80) and did not follow any consistent pattern.
When plotted as a function of net density (the metric generally most useful in compound networks for other phenotypes), no relationship was found for oscillation period of the W metric. This suggests that the oscillation period length is independent of the difference between the densities of activation and suppressive interactions. However, we found an interesting relationship between the total density (activator plus suppressor density) and W period. Above a total density of 0.4 (irrespective of the ratio of activators to suppressors), we found all possible dynamical profiles from stable fixed points to repetitive cycling, to complex oscillations with long cycling periods, including a few that appear to follow aperiodic and even chaotic dynamics. Periodicity at higher total densities thus appears to be entirely dependent on topological genotype, with no influence of density. Below DT=0.4, however, there was a clear exponential relation between total interaction density and period length, with no examples of very large period lengths or aperiodicity. Our data are consistent with the conclusion that extremely complex dynamical behavior is a function of very specific patterns of interaction within dense networks. No such patterns were readily discernible from examination of the topology of the aperiodic and chaotic networks, and further elucidation of the mathematical basis of such behavior will require more effort.
Very complex dynamic behavior has been extensively reported in biological GRNs and network models, including in certain regulatory motifs [37], in statistical analyses [15], and in systems of differential equations used to model such networks [16-18]. Payne et al. [19] used Random Boolean Circuits [55] to explore the effect of chaotic dynamics on robustness and evolvability.
In addition to supporting our hypothesis regarding the potential to find quantitative relationships to predict model dynamics when the effect of topological genotype is low, we have also presented a number of novel findings that could be important in future theoretical and applied studies into gene regulatory networks. We developed four equations, one each for the integrated values of HG0 and W for Activation and Compound networks. The two equations for Activation networks appear to be quite accurate with respect to the experimental simulation data and could be considered quantitative laws governing the dynamics of these networks. The equations for Compound networks provide the best available approximation of the experimental data but could still be useful in modeling real-world biological networks.
The high degree of functional complexity suggested by complex time-course behavior of these model interactive networks has some interesting implications for understanding how gene regulation might operate in biological systems. Compared to the situation in Boolean models (where genes can only be on or off, and they can only activate or suppress other genes), biological control of gene expression is far more complex and nuanced.
The use of randomly generated model GRNs in “experimental” studies of interactive network behavior is a valuable tool in the quest for useful quantitative relationships that might govern real-world networks, at least in approximation. Elucidation of such rules is likely to be useful not only in further studies of evolution and robustness of GRNs, but also in understanding the fundamental mechanisms by which interactive regulatory networks play such a central role in all aspects of biological function.
This work was supported by a Grant (#57657) from the John Templeton Foundation. The authors thank Mark Meredith, Kevin Rankine, and Emanuela Taioli