Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2008) Volume 1, Issue 1
In this study we report the first gel based proteomic analysis of an inflammed dry eye utilising a clinically-based non-invasive methodology for collection of a specimen from the posterior lid and inferior conjunctival mucosa of the subject. This multidimensional technique allowed the identification of 592 proteins, having a MOWSE score of greater than 40, using the heuristic tool PROVALT. Automated curation of this list using an inbuilt randomised database searching tool with false discovery rate set at 1% significantly reduced this list to 86 proteins. Additional manual curation resulted in the final positive identification of 75 proteins. These identified proteins were functionally classified and physiochemically characterised. This led to the identification of a number of proteins involved in cell structure, inflammation, and the innate immune response. Contained within these proteins were a number of potential biomarkers of not only dry eye syndrome but also lacrimal gland acinar cell function such as lacritin, calgranulin A and lacrimal proline-rich protein 4.
Keywords: Conjunctival swab; Dry eye; Proteomics
The front of the eye or ‘ocular surface’ comprises mucosal epithelium and various glandular structures that include lacrimal, accessory lacrimal and meibomian glands all of which are bathed in a thin fluid film. These structures are integrated together and function as a composite group designed to lubricate, nourish, protect and most importantly produce a clear, smooth optical surface to refract light (Stern et al.,1998; Rolando and Zierhut, 2001)
The overlying fluid tear film contains proteins, electrolytes and growth factors involved in maintaining the health of the ocular surface. The tear film is produced by lacrimal glands, meibomian glands, and the surface epithelium and is in close proximity to the systemic blood circulation via the highly vascularised submucosa. Various proteins within the tear film are recognised to play key roles in ocular defence including: lactoferrin, lysozyme, phospholipase A2, ceruloplasmin and immunoglobluins such as secretory IgA (Abe et al., 1999; Cullor et al., 1990; Glasson et al., 2002; Haynes et al., 1998; Haynes et al., 1999; O’Callaghan et al., 2003; Seal et al., 1986). Other defence proteins include trefoil peptides, SP-D (surfactant protein-D), gp340 (glycoprotein 340) and PLA2 and during inflammation the tear film quickly fills with inflammatory cells which leak from the adjacent blood vessels.
Recent research has demonstrated a plethora of proteins (>400) within the normal tear film (De Souza et al., 2006). The proteins present at any given time within the tears may be considered as a‘snapshot’ of the status of the ocular surface. Alterations in such proteins undoubtedly contribute to the dynamic of the ocular surface in health and disease. Previous examination of the tear film in dry eye conditions such as Sogrens have demonstrated alterations in key proteins such as lysozyme, lactoferrin and EGF (Seal et al., 1986; Ohashi et al., 2003; Schoenwald et al., 1998).
The purpose of this study was to analyse the applicability of a posterior lid margin, inferior fornix mucosal swab to profile proteins present within the tear film and ocular surface. Most sampling of the tear film to date has been through the use of capillary tear collection, a laborious and technically difficult approach with limited clinical applicability (Zhou et al., 2006). An extensive search of the literature revealed limited investigation of the protein profile present in a conjunctival sample taken from the inferior conjunctiva of human subjects (Grus et al., 2005; Tsai et al., 2006). The use of a simple non-invasive swab technique to include both tears and conjunctival cells would simplify this clinical test and produce a more complete proteomic characterisation of the ocular surface environment to include non secreted cellular proteins as well as those present within tear film. Recent advances in genomics and proteomics offer huge potential to enable clinicians to view biomarker patterns in both healthy and diseased ocular surfaces. This study was designed to investigate the proteome of an eye with dry eye syndrome clinically characterized by ocular surface inflammation, glandular dysfunction and an abnormal tear film.
Reagents
All reagents were purchased from Sigma-Aldrich (Poole, UK) with the exception of mass spectrometry grade water and acetonitrile, which were purchased from Romil (Cambridge, UK) and Trypsin, which was purchased from Promega (Southampton, UK).
Sample Collection and Patient Assessment
A patient who presented with dry eye symptoms was clinically assessed using a slit lamp examination of meibomian glands; lids, lid margins, conjunctiva and tear film;(Foulks and Bron, 2003) impression cytological assessment of conjunctival goblet cells; (Anshu et al., 2001; Saini et al., 1990) tear break-up time using fluorescein (Kojima et al., 2004) and the Zone-Quick phenol red thread test (PRT) (Menicon, USA) (Hamano et al., 1983). The subject also completed the McMonnies dry eye questi o nnaire (McMonnies et al., 1986).
Sample collection was performed in a clean ophthalmic consulting room and the examining ophthalmologist wore sterile gloves in order to minimise contamination of test samples. In addition, negative control swabs were taken at the time and place of subject testing to confirm the lack of environmental contamination.
After instillation of topical anaesthetic, the ocular specimen for protein analysis was collected from the posterior lid margin and lower conjunctival sac using a sterile cotton swab (Bibby Sterilin Ltd., Stone, UK). The swab was immediately cut into a sterile lysing matrix extraction tube containing ceramic and silica beads (BIO 101 Anachem, UK) and 1ml of sodium phosphate buffer using sterile scissors.
Protein Extraction and Quantification
The extraction tube containing the swab was processed using a FastPrep Instrument (Anachem, UK) at a preset speed of 6.0 for 30 s with a 2 min resting period on ice before the procedure was repeated. The sample was the centrifuged for 30 minutes at 25,000 g to remove cellular debris, the supernatant was decanted and stored frozen at –70°C until required. The total soluble protein content was measured using the Bradford assay (Bradford, 1976).
One Dimensional Gel Electrophoeresis
An aliquot of the supernatant (10 ìL) was added to 10ìL Tris-Glycine SDS sample loading buffer (Invitrogen, Renfrewshire, UK) and boiled for 5 min. The sample (20 ìL; 50 ìg total protein) was loaded onto a 1 mm thick Nu-Page 4-12 % Bis- Tris gel (Invitrogen, Renfrewshire, UK). SeeBlue™ Plus 2 (Invitrogen, Renfrewshire, UK) was used as a protein molecular mass marker. The gel was electrophoresed, using MES SDS running buffer, in an X-Cell II mini gel system (Invitrogen, Renfrewshire, UK) at 200 V, 120 mA, 25 W per gel for 35 min. Proteins were visualised using SimplyBlue™ Safestain (Invitrogen, Renfrewshire, UK). The entire lane was excised from the gel and cut into 3 mm fractions.
In-Gel Tryptic Digestion
Excised gel fractions were washed overnight in 200 ìL of a 50% (v/v) methanol and 5% (v/v) acetic acid solution. These fractions were then dehydrated by incubation for 5 min in 200 ìL acetonitrile. 30 ìL of 10 mM ditiothreitol was added and incubated for 30 mins at room temperature, followed by the addition of 30 ìL of 100 mM iodoacetamide for 30 min. 200 ìL acetonitrile was added and incubated for a further 5 min. Rehydration of gel fractions was carried out in 200 ìL of 100 mM NH4HCO3, pH 7.8 for 10 min at room temperature. Gel fractions were dehydrated as above usingacetonitrile and then dried in a rotary evaporator. 30 ?L of 20 ng/ìL trypsin in 50 mM NH4HCO3, pH 7.8 was added to each sample and incubated overnight at 37°C. 30 ìL of 50 mM NH4HCO3 was added to the samples and incubated for 10 min. The supernatant was subsequently recovered into microcentrifuge tubes and two further peptide extractions from these gel pieces were carried out with addition of 30 ìL 50% (v/v acetonitrile and 5% (v/v) formic acid) for 10 min. Peptide-containing liquid fractions were pooled, dried under vacuum and re-suspended in 20 μL 0.1% formic acid in 2% acetonitrile prior to storage at -70?C until required.
LC-MS Analysis
Mass spectrometry was performed using a 3200 Q-TRAP Hybrid ESI Quadropole linear ion trap mass spectrometer, ESI-Qq- Qlinear ion trap-MS/MS (Applied Biosystems/MDS SCIEX, Toronto, Canada) with a nanospray interface, coupled with an online Ultimate 3000 nanoflow liquid chromatography system (Dionex/LC Packings, Amsterdam, The Netherlands). A μ- Precolumn™ Cartridge (300 ìm × 5 mm, 5 ìm particle size) was placed prior to the C18 capillary column (75 μm × 150 mm, 3 μm particle size) to enable desalting and filtering. Both columns contained the reversed phase material PepMAP™ 100 (C18 silica-based) with a 100 Å pore size (Dionex/LC Packings). The elution buffers used in the gradient were Buffer A (0.1% formic acid in 2% acetonitrile) and Buffer B (0.1% formic acid in 80% acetonitrile). The nanoLC gradient used was 60 min in length: 0 – 55% B in 45 min, 10 min at 90% B followed by 5 min at 100% A. The flow rate of the gradient was 300 nLmin-1. The detector mass range was set at 400–1800 m/z. MS data acquisition was performed in positive ion mode. During MS acquisition peptides with 2+ and 3+ charge state were selected for fragmentation.
Database Searching, Protein Identification and PROVALT Analysis
Protein identification was carried out using an internal MASCOT server (version 1.9; Matrix Science, London, UK) searching against a human database extracted from the NCBI database (latest version at the time of processing). Peptide tolerance was set at ± 1.2 Da with MS/MS tolerance set at ± 0.6 Da and the search set to allow for 1 missed cleavage. Carbamidomethylation was set as a fixed modification and oxidation of methionine as a variable modification within the search parameter settings. In order to expedite the curation of the identified protein list from MASCOT, the result files were re-analysed against this extracted database using the heuristic method known as the protein validation tool PROVALT (Weatherly et al., 2005). This automated program takes large proteomic MS datasets and reorganises them by taking multiple MASCOT results and identifying those peptides that match. Redundant peptides are removed and related peptides are grouped together associated with their predicted matching protein, thus, the program dramatically reduces this portion of the curation process. For identification purposes the minimum peptide length was set at 6 amino acids, minimum peptide MOWSE score was set at 25 and the minimum high quality peptide MOWSE score was set at 40. PROVALT also uses peptide matches from a random database (in this case the extracted human database was randomised) to calculate falsediscovery rates (FDR) for protein identifications as previously described by Weatherly et al.(2005). Briefly, identifications from searching the normal and random databases are used to calculate the FDRs and set score thresholds and thus identify as many‘actual’ proteins as possible while encountering a minimal number of false-positive protein identifications. Rather than calculate error rates at the peptide level, the FDR calculations employed by PROVALT provide a reasonable balance between the number of correct and incorrect protein assignments. In this study the FDR was set at 1%, meaning that 99% of the reported proteins identified should be correct.
Eye Examination
Examination of the eye demonstrated an abnormal tear film, infla med ocular surface and abnormal lipid producing glands within the lid margins. Meibomian gland disease (MGD) was noted, characterised by significant plugging of more than 5 meibomian gland orifices, evidence of lid margin irregularity and inflammation, posterior meibomitis, palpebral and bulbar conjunctival inflammation and evidence of a mild capillary conjunctivitis. Tear volume as determined by the phenol red thread (PRT) test was not found to be significantly decreased. An overall reduced tear break-up time of 6 seconds, positive McMonnies questionnaire score of 18 (McMonnies et al., 1986), and presence of MGD indicated the presence of a dry eye syndrome secondary to abnormal tear film.
Comprehensive Analysis of Eye Swab Proteome
In this study we report the first gel based proteomic analysis of an inflammed dry eye specimen using a sterile cotton swab collected from the posterior lid and inferior conjunctival mucosa. This multidimensional analysis involved the eye swab proteome being first separated by one-dimensional gel electrophoresis. The resultant gel was then cut into 3 mm sections, each gel fraction was then trypsinized and the extracted peptides separated on a reversed phase C18 column over a 60 min time period prior to being introduced onto the mass spectrometer. This methodology allowed the identification of a total of 75 proteins (Table 1) from the eye swab utilising a false-discovery rate of 1% for protein detection.
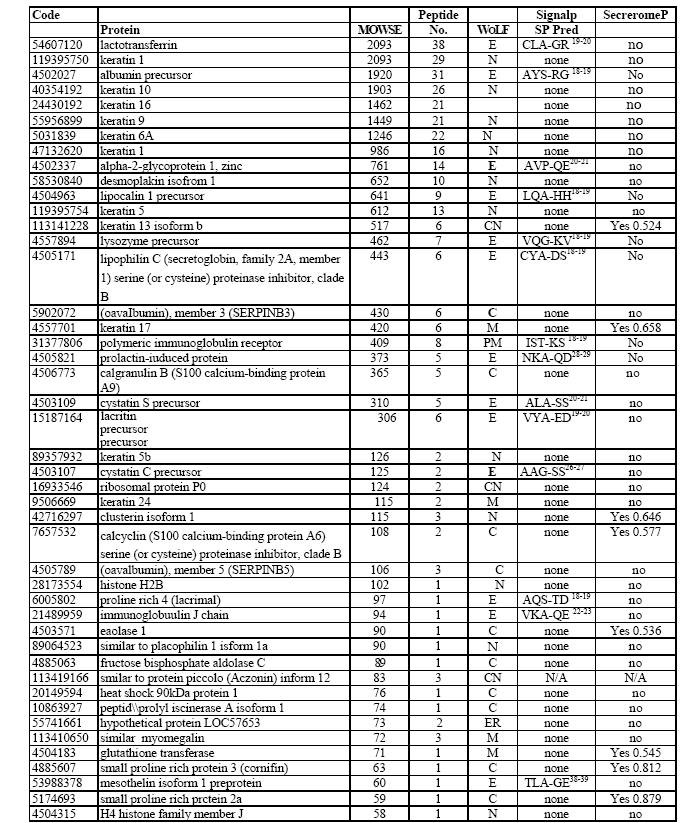
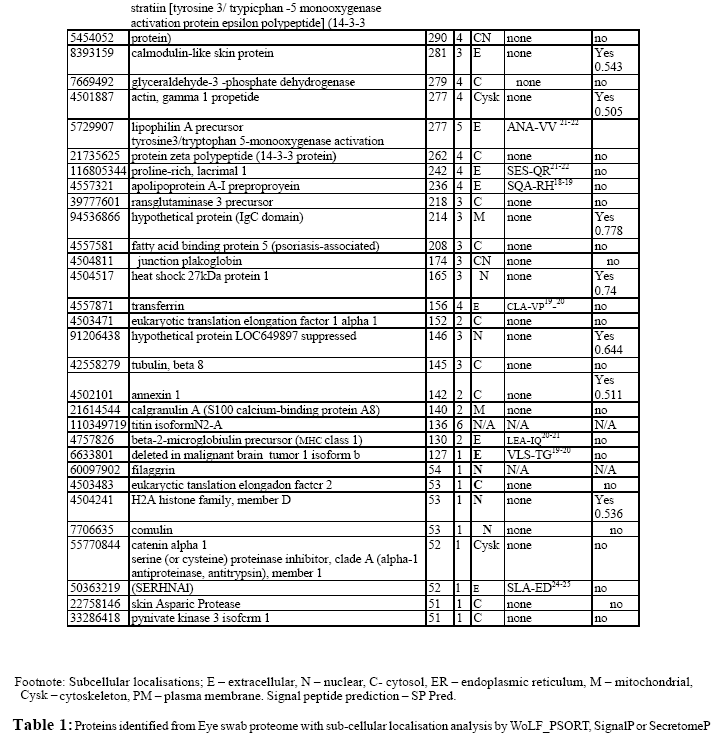
As previously reported, (Graham et al., 2006) due to the complex nature of the peptide mixtures to be analysed, the separation capabilities of the LC-MS systems are often exceeded. In this study all peptide fractions were analysed in duplicate in order to increase overall peptide identifications. In the current study, automated curation of our initial dataset, by the heuristic bioinformatic tool PROVALT utilising random database searching (Weatherly et al., 2005) led to the identification of 86 proteins with further manual curation leading to the positive identification of 75 proteins. The average number of peptides per protein was 6 and the average MOWSE score was 357, with the minimum high quality peptide MOWSE score for this study set at 40.
The 75 identified proteins had a wide range of physiochemical properties in respect to pI and molecular mass (Mr) (Figure 1). This 2-D visualisation showed that the smallest protein identified was small proline rich protein 2a (Mr = 7965 Da). The largest protein identified was titin isoform N2-A (Mr = 3713727 Da). The most acidic protein identified was calmodulin-like skin protein (pI = 4.43) while the most basic was H4 histone family member J (pI = 11.36).
Of the 75 proteins detected in this study, functional roles for 72 proteins (96 %) were known or could be predicted from database analysis. Proteins within the eye swab proteome were assigned to functional categories utilizing the bioinformatics tool ProteinFun (Jensen et al., 2003). Figure 2 shows that the largest category of identified proteins was cell envelope (32.4%), followed by those involved in translation (18.9%), then those involved in amino acid biosynthesis (13.5%). The remaining proteins were distributed amongst the other functional categories.

Figure 2: Chart showing the predicted functional categorisation of the proteins identified in the eye swab proteome utilising ProteinFun (Jensen et al., 2003).
The gene ontology of this protein dataset could also be identified utilizing ProteinFun (Figure 3) (Jensen et al., 2003). Of the 75 identified proteins over 38% had no prediction for gene ontology; however, 22% were identified as growth factors, 16% as stress response proteins and 7% as immune response proteins.

Figure 3: Chart showing the predicted ontology of the proteins identified in the eye swab proteome utilising ProteinFun (Jensen et al., 2003).
The rapid increase in genomic data over the past decade has revealed many important aspects of cellular processes, however there are still a significant number of potential gene products for which we know nothing, save that they are classified as ‘hypothetical proteins’. In previous work we have underlined the necessity to assign, where possible, an element of biological functionality to such gene products in order to develop both systems biology and our understanding of cellular processes within the system under investigation. Within the current study we have established the presence of three proteins that had previously been annotated as hypothetical conserved proteins (Table 1). The identification of such proteins establishes the biological functionality of these ‘hypothetical’ predicted protein coding sequences, and elegantly demonstrates the potential of proteomics to validate bioinformatics predictions.
Having established the presence of such proteins and wishing to understand how they contribute to functional processes we further examined them using NCBI BLASTp. Such an approach allows conserved domains within protein sequences to be identified and thereby enables a degree of inferred functionality. Using this methodology allowed us to assign putative function to one of these proteins, hypothetical protein LOC649897. This protein contained two IGc domains (CD00098) these are part of the immunoglobulin domain constant region subfamily; members of the IGc subfamily are components of immunoglobulins, T-cell receptors, CD1 cell surface glycoproteins, secretory glycoproteins A/ C, and Major Histocompatibility Complex (MHC) class I/II molecules. This once again demonstrates the ability of proteomics to investigate and validate genomic analyses studies.
Sub-cellular Protein Localisation
Sub-cellular localization prediction tools have been used for many years to identify those proteins that are retained by and exported from cells. They may also have uses in identifying possible diagnostic and therapeutic targets as well providing information on the functionality of a protein (Gardy et al., 2005). In the current study a number of bioinformatics tools including WoLF_PSORT (Horton et al., 2007), SignalP (Bendtsen et al., 2004) and SecretomeP (Bendtsen et al., 2005) were utilized. These bioinformatics tools endeavour to assign a sub-cellular location for each protein. These tools use a set of descriptor rules and a variety of computational algorithms and networks to analyse a proteins’ amino acid composition in an attempt to identify known motifs or cleavage sites.
All 75 proteins identified in this study were initially analysed using WoLF_PSORT; 20 were predicted to be cytoplasmic, 21 proteins were predicted to be extracellular, 18 were predicted to be nuclear with the remaining 16 having other sub-cellular localisations (Figure 4). These protein subsets were further analysed using SignalP, to predict amino-terminal signal peptides, and SecretomeP, which attempts to identify non-classically secreted proteins. Of those 20 proteins classified by WoLF_PSORT as being cytoplasmic, 15 were confirmed as non-secretory by this new analysis, while 5 were predicted to be potential secretory proteins (all non-classically secreted). Of the 21 proteins initially predicted by WoLF_PSORT to be extracellular all were predicted to be secreted, 20 had predicted signal peptide sequences, one was identified as non-classically secreted. Of the 18 proteins identified by initial analysis as nuclear, 13 were confirmed as nonsecretory proteins, 4 were predicted to be non-classically secreted and one had no information available. Of those 16 proteins predicted to have localizations other than the three above, eight were predicted to be non-secretory, six were predicted to be secreted (5 non-classically) and two had no information available.

Figure 4: Overview of bioinformatics analysis of identified proteins from the eye swab proteome. Cellular localisation was predicted based upon the use of WoLF_PSORT (Horton et al., 2007), SignalP v3.0 (Bendsten et al., 2004) and SecretomeP v2.0 (Bendsten et al.,2004).
The 21 proteins identified, from the above analyses, as possessing an N-terminal signal peptide were grouped and analysed in order to ascertain whether they have the required domain architecture for eukaryotic signal peptides; namely an N-terminal region (n-region) that usually has a net positive charge, a hydrophobic core (h-region) containing six to fifteen hydrophobic amino acids and a C- terminal (c-region) which often contains helix breaking proline or glycine and small uncharged residues in position – 3 and -1 (Table 2). As can be seen all the proteins predicted to contain signal peptides and therefore be secreted do indeed have the required architecture (Martoglio and Dobberstein, 1998).

Protein Characterization
A goal of the current study was to ascertain the potential of this clinical sampling technique for the characterisation of dry eye syndrome proteome. As such it would be expected that our data set would incorporate proteins associated with tears and the ocular surface, whose functions would include roles in innate immunity, inflammation response and of course general cellular functions. Below we describe the categories of proteins detected in the current investigation outlining their cellular role and potential as biomarkers for dry eye syndrome.
A number of cytoskeletal structural proteins could be identified including micro- and intermediate filaments (keratins, filaggrin, actin, titin); microtubules (tubulin); catenins (plakoglobin) and other cytoskeletal proteins (desmoplakin, myomegalin) (Table 1). Myomegalin is a novel protein that has been found to act as an anchor to localise components of the cAMP-dependent pathway to the Golgi/centrosomal region of the cell (Verde et al., 2001). Whilst keratin is known to be a contaminant in many proteomic investigations its presence here is not unexpected with Tsai et al., (2006) reporting some 10 keratin isoforms in their investigation of meibomian gland secretions. It is likely that the origin of keratin within the sample is the skin surrounding the eye, some of which will have been ‘harvested’ during eye swab preparation. A recent study of the tear fluid proteome by de Souza et al., (2006) identified all of these cytoskeletal structural proteins with the exception of keratin isoforms, filaggrin and myomegalin.
Four proline-rich proteins (PRPs) (Table 1) were also identified in this study. The biological importance of PRPs in the eye lies in their antimicrobial properties (Zhou et al., 2006). Fung et al., (2004) proposed that PRPs play a significant role in protection of the ocular surface and are involved in pathogenesis of inflammatory and autoimmune diseases; whilst Grus et al., (2005) implicated PRPs in the modulation of the eye microflora. The significance of PRPs in eye health diagnosis was also highlighted by Grus and coworkers (Grus et al., 2005) who proposed that lacrimal prolinerich protein 4, also detected in the current study, would be a useful biomarker for human lacrimal gland acinar cell function.
Within this investigation a number of calcium binding proteins were observed. These proteins are known to have a role in the innate immune response for example annexin is an important mediator of the anti-inflammatory actions of glucocorticoids, and calmodulin is also involved in inflammation response (Table 1) (Buckingham et al., 2006). In addition calprotectin (which is a complex of calgranulin A and B) was also identified; this myeloidrelated protein complex is known to possess in vitro bacteriostatic and fungistatic properties and is found at high abundance in neutrophils, cells that are rapidly attracted to sites of inflammation (Herndon et al., 2003; Gaya and Mackenzie, 2002). Calprotectin is therefore a potential biomarker for a range of inflammatory disease states such as Crohns’ disease (Gaya and Mackenzie, 2002). Indeed Grus et al., (2005) identified increased abundance of calgranulin A in dry eye patients and proposed this protein as a useful indicator for this condition.
It has previously been shown (De Souza et al., 2006) that the proteome of tear fluid contains a number of proteins with protease inhibiting properties which was confirmed in our eye swab sample (Table 1). Such proteins included lipocalin 1 which in addition to its protease inhibiting activity, is known to bind a diverse range of molecules, functioning as a scavenger of physiologically damaging lipophilic ligands (Tsai et al., 2006). Cystatin C and S are extracellular proteins belonging to the family 2 cystatins that are ubiquitous in all human secretions and thought to be part of a non-immune protective system, functioning by inhibition of extra- or intracellular microbial cysteine proteases (Jasir et al., 2003).
Three serine proteinase inhibitors (SERPINs) were also identified in this study. SERPINA1 (alpha 1 antitrypsin) controls the activity of a diverse range of proteolytic enzymes and plays an important role in infection control by inactivating enzymes activated by bacteria and has been shown to play a role in regulating immune response by inhibiting the migration and transformation of lymphocytes (Sen et al., 1988). SERPINB3 and SERPINB5 are members of the Ov-serpins and have been shown to inhibit a number of proteinases. Their presence in epithelial cells is suggestive of a role in barrier/host defence against microbial or viral proteinases (Silverman et al., 2001).
It has been reported that 35-45% of normal human tears is comprised of a number of proteins with antimicrobial properties that form part of the innate immune response including lysozyme, lactotransferrin, transferrin, and IgA (Lehrer et al., 1998). Lysozyme is an enzyme present in human neutrophils, whose bacteriocidal activity is precipitated via the degradation of peptidoglycan, a major component of bacterial cell walls (Laible and Germaine, 1985). Transferrin and lactotransferrin interfere in microbial growth by effectively binding iron (Arnold et al., 1977; Arnold et al., 1982). Lactotransferrin is highly abundant in granules of human neutrophils and secretions from epithelial cells and can also be directly microbiocidal (Arnold et al., 1982). Both lactotransferrin and lysozyme are known to function as opsonins, effectively ‘marking’ the bacterial cells for phagocytosis by white blood cells (Jenssen, 2005). Lactotransferrin and lysozyme have also been shown to have synergistic activitiy (Travis et al., 1999). Secretory IgA is considered the first line of immune defense (Kaetzel et al., 1991) and has been reported to be present in those few tear proteomic investigations carried out to date (Zhou et al., 2006; Tsai et al., 2006). It is surprising therefore that IgA was not detected in the current study, however the polymeric immunoglobulin receptor (pIgR) which mediates the transfer of secretory IgA into external fluids was identified. In addition IgJ, the synthesis of which is critical for primary immune response, due to its function in assembling both IgA and IgM, was also present (Matsuuchi et al., 1986; Iwata et al., 2002). A possible explanation for our failure to detect IgA may be explained by the dimeric/polymeric nature of IgA during the immune response leading to the formation of a very high molecular weight protein that may be intractable by our gel-based method of analysis (Stubbe et al., 2000).
Also present within the swab sample was lipophilin A and C which are known to form a heterodimeric molecule in vivo. Whilst lipophilin C has been reported to have weak antimicrobial properties none have been recorded so far for lipophilin A. Indeed at present the role of this lipophilin heterodimer is unknown however it is part of the uteroglobin superfamily and has been found to be capable of transporting steroid and other nonpolar molecules. The ubiquity of lipophilins within human secretions suggests that they are likely to serve important functions in many different tissues and organs (Lehrer et al., 1998; Lehrer et al., 2000). A final protein detected in the swab that possessed antimicrobial properties was prolactin-induced protein (PIP) which binds to and thereby inhibits the growth of certain bacteria, such as Streptococcus (Schenkels et al., 1997). In addition to the antimicrobial role of PIP in secreted fluids it is also known to be a potent inhibitor of T lymphocyte programmed cell death induced by crosslinking of CD4 and T cell receptor (Gaubin et al., 1999; Kitano et al., 2006).
A number of proteins associated with inflammation and other disease processes were found (Table 1; apolipoprotein A1 (Burger and Dayer, 2002); alpha 2 glycoprotein 1 (Hale and Price, 2001); and DMBT1 (Mollenhauer et al., 2000). The protein lacritin, which was also detected is normally found in tear films and is known to stimulate new tear production but has also been shown to be deficient in dry eye syndrome and so may act as a useful biomarker for this disease (Koo et al., 2005).
This proteomic study of a dry eye patient demonstrates the variety of known and unknown proteins/peptides present in this pathological state. This study highlights the possibility of using a simple non-invasive clinical swab technique in these patients rather than more complex clinical testing procedures in order to investigate dry eye syndrome and potentially other disease states of the eye. Further research is required to fully characterise relationships between clinical signs/symptoms and the proteome. The determination of protein changes which imply pathology and/or which are harbingers of pathology will enable focussed therapeutic treatments and may enable prophylaxis against dry eye conditions.
*The work of J Graham was funded under a Northern Ireland‘Proof of concept’ grant.
R. L. J Graham was supported by the Northern Ireland Centre for Excellence in Functional Genomics, with funding from the European Union (EU) Programme for Peace and Reconciliation, under the Technology Support for the Knowledge-Based Economy.