Journal of Theoretical & Computational Science
Open Access
ISSN: 2376-130X
ISSN: 2376-130X
Research Article - (2015) Volume 2, Issue 3
Quantum chemical calculation of geometries and vibrational wavenumbers of 2-(1-piperazinyl) ethanol in a ground state level are carried out by using density functional theory (DFT/B3LYP) method with 6-31+G(d,p)basis set. The harmonic vibrational frequencies are calculated and scaled values have been compared with experimental FTIR, FT-RAMAN spectra. The stability of the molecule arising from hyper conjugative interaction and the charge delocalization has been analyzed using natural bond orbital (NBO) analysis. A study on the electronic properties such as HOMO and LUMO energies are performed by time-dependent DFT (TD-DFT). NLO property of the title compound is calculated. The 1H and 13C NMR chemical shifts of the molecule are calculated by the gauge independent atomic orbitals method. The mapping of electrostatic potential energy surface (MEP) is performed for the title compound.
<Keywords: Vibrational analysis; UV; NBO; NMR; HOMO-LUMO
2-(1-Piperazinyl) ethanol has a general formula C6H14N2O which belongs to the family of piperazine. Molecular weight of 2-(1-piperazinyl) ethanol 130.18 g/mol. Piperazine derivatives are widely used in pharmacological and electro optic applications. The piperazine template forms the molecular backbone; possess versatile binding properties with a frequency occurring binding motif which provides potent and selective ligands for a range of different biological targets in medicinal chemistry. Piperazines are currently the most important building blocks in drug discovery, with a high number of positive hits encountered in biological screens of this heterocyclic and its congeners across a number of different therapeutic areas [1,2]; anticancer [3-5], antifungal [6], antibacterial, antimalarial and antipsychotic agents [7], as well as HIV protease inhibitors [8,9]. Piperazines kill tumor cells directly through the induction of apoptosis. Their anti-tumour modes of action are quite distinct and are significantly more potent. They are active against a variety of different tumor types and orally bioavailable [10] piperazine derivatives can act as stimulants at low doses, while causing hallucinations at higher doses [11] and are designed as designer drugs.
To the best of our knowledge, neither quantum chemical calculations, nor vibrational analysis study of 2-(1-piperazinyl) ethanol has not been reported yet. DFT technique is employed to study the complete vibrational spectra of the title compound and to identify the various normal modes with greater wave number accuracy. Therefore, in the present study, FTIR and FT-RAMAN spectral analysis of 2-(1-piperazinyl) ethanol, have been recorded and verified by using density functional theory (DFT). UV and NMR studies are also carried out by B3LYP /6-31+G(d,p) basis set. The redistribution of electron density (ED) in various bonding, anti-bonding and E2 energies had been calculated by natural bonding orbital (NBO) analysis to give clear evidence of stabilization originating from the hyper conjugation of various intra molecular interactions. The HOMO and LUMO analysis have been used to elucidate information regarding charge transfer within the molecule.
The sample of the present compound 2-(1-piperazinyl) ethanol was purchased from Merck chemical company, with spectroscopic grade and it is used as such without further purification. The FT-IR spectrum of the compound has been recorded in Perkin-Elmer spectrometer between 4000 and 400 cm-1. The spectral resolution is ± 1 cm-1. The FT-Raman spectrum of the compound is also recorded in the same instrument with FRA 106 Raman module equipped with Nd: YAG laser source operating at 1.064 μm line widths with 200 mW powers. The spectra are recorded with scanning speed of 30 cm-1. The frequencies of all sharp bands are accurate to ± 1 cm-1. 1H and13C nuclear magnetic resonance (NMR)(400 MHz; CDCl3) spectra were recorded on a Bruker HC instrument. Chemical shifts for protons are reported in parts per million scales (δ scale) downfield from tetramethylsilane (TMS).
Quantum chemical calculations
Calculations of the title compound were carried out with Gaussian 09 software program [12] using the B3LYP/6-31G+(d,p) to predict the molecular structure and vibrational wave numbers. Calculations were carried out with Beck’s three parameter hybrid model using the Lee-Yang-Parr correlation functional (B3LYP) method. The structure optimization was performed to confirm the structure and hence to find the optimized geometry of the examined species, no imaginary wave number modes were obtained by B3LYP/6-31+G(d,p). The wavenumbers values computed contain known systematic errors and therefore, have used the constant scaling factor value of 0.956 for DFT method [13]. The DFT hybrid B3LYP functional method tends to overestimate the wavenumbers of functional modes; therefore scaling factors have to be used for obtaining a considerably better agreement with experimental data [13]. The obtained geometrical parameters (B3LYP) are given in Table 1. The assignment of the calculated wave numbers are aided by the animation option of Gauss view program, which gives a visual presentation of the vibrational modes [14].
| Parameters | Theoritical bond length (Å) | |
|---|---|---|
| C1-H2 | 1.0957 | |
| C1-H3 | 1.0973 | |
| C1-C4 | 1.5362 | |
| C1-N13 | 1.466 | |
| C4-H5 | 1.0971 | |
| C4-H6 | 1.1104 | |
| C4-N15 | 1.4614 | |
| C7-H8 | 1.1104 | |
| C7-H9 | 1.097 | |
| C7-C10 | 1.5361 | |
| C7-N15 | 1.4614 | |
| C10-H11 | 1.0973 | |
| C10-H12 | 1.0957 | |
| C10-N13 | 1.466 | |
| N13-H14 | 1.0184 | |
| N15-C16 | 1.459 | |
| C16-H17 | 1.0967 | |
| C16-H18 | 1.0967 | |
| C16-C19 | 1.5364 | |
| C19-H20 | 1.0994 | |
| C19-H21 | 1.0994 | |
| C19-O22 | 1.4304 | |
| O22-H23 | 0.9651 | |
| Bond angle (Å) | ||
| H2-C1-H3 | 107.7412 | |
| H2-C1-C4 | 110.1545 | |
| H2-C1-N13 | 108.7599 | |
| H3-C1-C4 | 108.7978 | |
| H3-C1-N13 | 107.8154 | |
| C4-C1-N13 | 113.397 | |
| C1-C4-H5 | 110.0597 | |
| C1-C4-H6 | 108.6202 | |
| C1-C4-N15 | 109.8313 | |
| H5-C4-H6 | 107.343 | |
| H5-C4-N15 | 108.6565 | |
| H6-C4-N15 | 112.2923 | |
| H8-C7-H9 | 107.3428 | |
| H8-C7-C10 | 108.6196 | |
| H8-C7-N15 | 112.2912 | |
| H9-C7-C10 | 110.0631 | |
| H9-C7-N15 | 108.6577 | |
| C10-C7-N15 | 109.8287 | |
| C7-C10-H11 | 108.7871 | |
| C7-C10-H12 | 110.1583 | |
| C7-C10-N13 | 113.3937 | |
| H11-C10-H12 | 107.742 | |
| H11-C10-N13 | 107.8138 | |
| H12-C10-N13 | 108.7611 | |
| C1-N13-C10 | 111.3619 | |
| C1-N13-H14 | 109.781 | |
| C10-N13-H14 | 109.7811 | |
| C4-N15-C7 | 111.3619 | |
| C4-N15-C16 | 115.437 | |
| C7-N15-C16 | 115.4382 | |
| `N15-C16-H17 | 108.3226 | |
| N15-C16-H18 | 108.3198 | |
| N15-C16-C19 | 116.9573 | |
| H17-C16-H18 | 106.8048 | |
| H17-C16-C19 | 108.0033 | |
| H18-C16-C19 | 107.9999 | |
| C16-C19-H20 | 110.5693 | |
| C16-C19-H21 | 110.5696 | |
| C16-C19-O22 | 106.3501 | |
| H20-C19-H21 | 108.134 | |
| H20-C19-O22 | 110.6175 | |
| H21-C19-O22 | 110.6175 | |
| C19-O22-H23 | 109.3781 | |
| H2-C1-C4-H5 | 64.1556 | |
| H2-C1-C4-H6 | -53.0962 | |
| H2-C1-C4-N15 | -176.2596 | |
| H3-C1-C4-H5 | -53.7368 | |
| H3-C1-C4-H6 | -170.9887 | |
| H3-C1-C4-N15 | 65.8479 | |
| N13-C1-C4-H5 | -173.6985 | |
| N13-C1-C4-H6 | 69.0497 | |
| N13-C1-C4-N15 | -54.1138 | |
| H2-C1-N13-C10 | 173.5921 | |
| H2-C1-N13-H14 | 51.7992 | |
| H3-C1-N13-C10 | -69.8454 | |
| H3-C1-N13-H14 | 168.3617 | |
| C4-C1-N13-C10 | 50.6738 | |
| C4-C1-N13-H14 | -71.1191 | |
| C1-C4-N15-C7 | 57.74 | |
| C1-C4-N15-C16 | -167.5245 | |
| H5-C4-N15-C7 | 178.1783 | |
| H5-C4-N15-C16 | -47.0862 | |
| H6-C4-N15-C7 | -63.2369 | |
| H6-C4-N15-C16 | 71.4987 | |
| H8-C7-C10-H11 | 171.0103 | |
| H8-C7-C10-H12 | 53.1152 | |
| H8-C7-C10-N13 | -69.0327 | |
| H9-C7-C10-H11 | 53.7572 | |
| H9-C7-C10-H12 | -64.1379 | |
| H9-C7-C10-N13 | 173.7142 | |
| N15-C7-C10-H11 | -65.8296 | |
| N15-C7-C10-H12 | 176.2753 | |
| N15-C7-C10-N13 | 54.1275 | |
| H8-C7-N15-C4 | 63.2266 | |
| H8-C7-N15-C16 | -71.5084 | |
| H9-C7-N15-C4 | -178.1888 | |
| H9-C7-N15-C16 | 47.0763 | |
| C10-C7-N15-C4 | -57.7471 | |
| C10-C7-N15-C16 | 167.5179 | |
| C7-C10-N13-C1 | -50.6807 | |
| C7-C10-N13-H14 | 71.1122 | |
| H11-C10-N13-C1 | 69.8345 | |
| H11-C10-N13-H14 | -168.3727 | |
| H12-C10-N13-C1 | -173.6024 | |
| H12-C10-N13-H14 | -51.8096 | |
| C4-N15-C16-H17 | 55.6992 | |
| C4-N15-C16-H18 | 171.196 | |
| C4-N15-C16-C19 | -66.5558 | |
| C7-N15-C16-H17 | -171.1758 | |
| C7-N15-C16-H18 | -55.679 | |
| C7-N15-C16-C19 | 66.5692 | |
| N15-C16-C19-H20 | -59.8388 | |
| N15-C16-C19-H21 | 59.89 | |
| N15-C16-C19-O22 | -179.9744 | |
| H17-C16-C19- H20 | 177.7407 | |
| H17-C16-C19-H21 | -62.5305 | |
| H17-C16-C19-O22 | 57.6051 | |
| H18-C16-C19-H20 | 62.5752 | |
| H18-C16-C19-H21 | -177.696 | |
| H18-C16-C19-O22 | -57.5604 | |
| C16-C19-O22-H23 | -179.9632 | |
| H20-C19-O22-H23 | 59.9325 | |
| H21-C19-O22-H23 | -59.8585 | |
Table 1: Optimized geometrical parameters of 2-(1-piperazinyl) ethanol by B3LYP/6-31G+(d,p).
Molecular geometry
The stability of the molecule was studied with the help of conformational analysis. Two conformations gauche and anti with respect to the C16-C19 has been carried out. The global minimum energy was found to be for the most stable conformer -421.76758126 Hartee (anti). The optimized geometrical parameters namely bond lengths, bond angles and dihedral angles are calculated by B3LYP/6- 31+G(d,p) which are listed in Table 1. The molecular structure of 2-(1-piperazinyl) ethanol with the atoms numbering are shown in the Figure 1.

Figure 1: Molecular Structure of 2-(1-piperazinyl) ethanol with atom numbering scheme.
From the table, C-H bonds are having the bond lengths in the range of 1.0957 Å to 1.1104 Å. The presence of the ethanol group attached to the N15 atom has shifted the bond distances of C4-H6 , C7-H8 to higher value (1.1104 Å) The bond lengths of C1-H3 , C7-H9, C10-H9 and C10-H11 are greater than the other C-H bonds lengths, shows that the electronegative atoms N13 is nearly to those bonds. The C–C bond values are ranging from 1.5361 Å to 1.5364 Å. The highly electro negative oxygen atom distorted the angle C16- C19-H20 and C16-C19-H21 to higher angle (110.56°).
The steric hindrance on N atom and column interaction between the H atoms of CH2 and CH3 groups give rise to the lone pair to be oriented in axial position while an ethanol group stays at equatorial position. The N-C bond lengths are found in the range of 1.45 Å to 1.465 Å which is comparing shorter than the experimental N-C bond length. This is due to the conjugation of its electro negativity character of the atom O22 which is present in the ethanol group. The N-H bond length takes the value 1.0184 Å. The other O-H and C-O bond lengths are found at 0.965 Å and 1.4304 Å respectively.
Piperazine can have different conformations which are chair, halfchair, boat, twist boat and envelope forms. Chair conformation was found to be the most stable conformer and Hendrickson proposed that for chair–chair inter conversion the most stable transition state would be one of the possible chair forms [15-17]. The dihedral angle calculated for C1-C4-N15-C16 is found to be -167.5245. Thus, the obtained optimized geometrical parameters from the Table 1 confirm the chair conformation of 2-(1-piperazinyl) ethanol.
Vibrational analysis
The aim of the vibrational analysis is to find the vibrational modes connected with calculated specific molecular structure of the compound. The molecule consists of 23 atoms which undergo 63 normal modes of vibration. The title molecule belongs to C1 point group symmetry. Vibrational band assignments of 2-(1-piperazinyl) ethanol have been made by using Gauss view molecular visualization program [18]. After applying the scale factors, the theoretical calculations reproduce the experimental data which are good agreement with literature value. The observed and scaled theoretical frequencies, IR intensities, Raman activities and normal modes of vibrations are listed in Table 2. The theoretical and experimental FTIR and FT-RAMAN spectrum are shown in the Figure 2.
| S.No | Observed frequencies | Calculated frequencies(*) | IR intensity | Raman | PED (%) | ||
|---|---|---|---|---|---|---|---|
| IR | Raman | Unscaled IR | Scaled IR | ||||
| 1 | 70 | 67 | 10.91 | 0.04 | τOCHH(63), τHOCC(11) | ||
| 2 | 95 | 91 | 1.72 | 0.33 | τCCNC(62), τHCNC(18) | ||
| 3 | 102 | 97 | 0.42 | 0.32 | τOCCN(58), τHOCC(13) | ||
| 4 | 228 | 218 | 2.43 | 0.26 | τCCNH(33), τHCNC(18) | ||
| 5 | 254 | 243 | 8.25 | 0.29 | δCCNC(43), τHCNC(14) | ||
| 6 | 258 | 247 | 114.91 | 1.68 | τHOCC(87), τOCHH(10) | ||
| 7 | 312 | 298. | 6.12 | 2.10 | τOCCN(50), τHOCC(30) | ||
| 8 | 329 | 336 | 321 | 15.51 | 0.67 | τ HCNC(44), τHHCN(13) | |
| 9 | 371 | 355 | 0.22 | 0.18 | τ HHCN(58), τHCNC(28) | ||
| 10 | 400 | 383 | 11.91 | 1.48 | τHCNC(47), τHCCH(10) | ||
| 11 | 460 | 483 | 492 | 471 | 3.02 | 2.38 | τHCCN(54), τHCCH(23) |
| 12 | 524 | 500 | 0.90 | 0.59 | τCCNH(20), τHCCH(12) | ||
| 13 | 585 | 641 | 613 | 35.85 | 1.45 | δCCNC(27), τHCNC(9) | |
| 14 | 773 | 739 | 40.85 | 11.61 | τCNHC(44), τHCNC(14) | ||
| 15 | 790 | 755 | 100.70 | 4.54 | τCNCH(42), τHCNC(20) | ||
| 16 | 769 | 799 | 764 | 0.16 | 0.52 | τHCCH(54), τCCNC(17) | |
| 17 | 847 | 810 | 0.19 | 1.18 | τ CNCC(17), τHHCN(13) | ||
| 18 | 852 | 896 | 857 | 11.81 | 4.47 | τHNCC(13), τHHCN(11) | |
| 19 | 879 | 911 | 871 | 2.78 | 0.10 | βCCN(38), νCC(19) , βCCH(10) | |
| 20 | 936 | 985 | 942 | 44.13 | 8.78 | βCCN(65), νCC(30) | |
| 21 | 1011 | 967 | 28.66 | 7.06 | νCC(61), νCN(22), βCH(10) | ||
| 22 | 1038 | 993 | 2.84 | 0.34 | τHCNC(20), τHHCN(10) | ||
| 23 | 1041 | 995 | 10.93 | 1.87 | τHCNC(22), τHHCN(13) | ||
| 24 | 1011 | 1061 | 1014 | 68.32 | 10.20 | νCO(80), νCN(15) | |
| 25 | 1057 | 1035 | 1091 | 1043 | 11.94 | 2.20 | νCN(73), βCH(22) |
| 26 | 1065 | 1066 | 1144 | 1094 | 12.10 | 2.55 | νCN(70), βCH(13) |
| 27 | 1144 | 1094 | 30.12 | 0.58 | νCN(60), νCC(17), βCH(13) | ||
| 28 | 1120 | 1182 | 1130 | 80.05 | 6.40 | νCN(38) , βHCH(18), βCOH(11) | |
| 29 | 1142 | 1193 | 1140 | 9.98 | 1.36 | νCN(41), βHCN(13) | |
| 30 | 1182 | 1212 | 1158 | 6.48 | 5.30 | βHCN(32), νCN(14) | |
| 31 | 1196 | 1224 | 1170 | 21.09 | 7.23 | βCOH(60), βHCH(28), νCN(09) | |
| 32 | 1202 | 1199 | 1246 | 1191 | 0.68 | 0.98 | νCC(53) , βCCN(18), βHCH(12) |
| 33 | 1303 | 1246 | 5.06 | 8.74 | βCCH(48), νCN(22) | ||
| 34 | 1268 | 1327 | 1269 | 7.36 | 3.83 | βCCN(59), νCC(18) | |
| 35 | 1286 | 1350 | 1291 | 1.72 | 10.9 | βHCN(69), βCNC(28) | |
| 36 | 1352 | 1290 | 19.58 | 4.14 | βCCN(28), βCNC(10) | ||
| 37 | 1297 | 1299 | 1356 | 1296 | 7.28 | 0.46 | βHCC(58), βHOH(22) |
| 38 | 1306 | 1365 | 1305 | 4.88 | 1.88 | βHOC(50) | |
| 39 | 1321 | 1389 | 1328 | 1.40 | 1.40 | βHCC(61) | |
| 40 | 1339 | 1396 | 1334 | 8.31 | 3.58 | βHCC(40), νCC(20) , νCN(13) | |
| 41 | 1364 | 1416 | 1353 | 7.07 | 2.22 | νCC(63), βCCN(20) | |
| 42 | 1450 | 1386 | 0.81 | 1.47 | βCOH(51), βHCH(13) | ||
| 43 | 1407 | 1477 | 1412 | 0.25 | 20.23 | βHCH(79) | |
| 44 | 1480 | 1415 | 8.89 | 3.89 | βHNC(47), βHCH(38) | ||
| 45 | 1484 | 1418 | 8.37 | 3.79 | βHCH RING +ETHANOL(80) | ||
| 46 | 1488 | 1422 | 4.25 | 11.60 | β HCH RING +ethanol(78) | ||
| 47 | 1497 | 1431 | 2.72 | 3.58 | βHCH(57), βNHC(21) | ||
| 48 | 1445 | 1447 | 1503 | 1437 | 12.00 | 2.25 | βHCH ring+ethanol(79) |
| 49 | 1466 | 1466 | 1529 | 1467 | 3.27 | 3.71 | βHCH ethanol(80) |
| 50 | 2746 | 2882 | 2756 | 33.66 | 22.59 | νCH(85) | |
| 51 | 2892 | 2765 | 145.72 | 153.17 | νCH(83) | ||
| 52 | 2879 | 3002 | 2870 | 49.82 | 112.65 | νCH(78) | |
| 53 | 3035 | 29O2 | 56.89 | 88.98 | νCH (88) | ||
| 54 | 3036 | 2903 | 5.04 | 32.75 | νCH(90) | ||
| 55 | 3039 | 2905 | 36.3817 | 170.3565 | νCH(95) | ||
| 56 | 3041 | 2907 | 43.7761 | 48.6382 | νCH(92) | ||
| 57 | 2917 | 3059 | 2924 | 74.6573 | 50.1508 | νCH(93) | |
| 58 | 3061 | 2924 | 32.5528 | 250.5377 | νCH(96) | ||
| 59 | 2946 | 2947 | 3089 | 2953 | 7.3905 | 122.6584 | νCH(98) |
| 60 | 3089 | 2953 | 46.2799 | 112.7917 | νCH(98) | ||
| 61 | 3088 | 2954 | 66.5198 | 10.9260 | νCH(99) | ||
| 62 | 3382 | 3504 | 3350 | 0.6450 | 73.0198 | νNH(100) | |
| 63 | 3837 | 366 | 30.0620 | 164.2640 | νOH(100) | ||
(*) –The frequencies calculated for the most stable conformer at the B3LYP/6-31+(d,p)
Table 2: Observed and calculated vibrational frequencies of 2-(1-piperazinyl) ethanol by B3LYP/631+G (d,P).

Figure 2: Comparison of experimental and calculated FTIR and FTraman spectrum of 2-(1-piperazinyl) ethanol.
CH2 vibrations: For the assignments of CH2 group frequencies, basically six fundamentals can be associated with each CH2 group namely, CH2 symmetric stretch; CH2 asymmetric stretch; CH2 scissoring and CH2 rocking, which belongs to in-plane vibrations and two out-ofplane vibrations, viz., CH2 wagging and CH2 twisting modes, which are expected to be polarized. The asymmetric CH2 stretching vibrations are generally observed above 3000 cm-1, while the symmetric stretch will appear between 3000 and 2900 cm-1 [19].
The hetero aromatic compounds and its derivatives are structurally very close to benzene. The C-H stretching vibrations of the aromatic and hetero aromatic structures occur in the region 2800-3100 cm-1. This permits the ready identification of the structure. The C-H stretching vibrations are identified at 2954, 2953, 2924, 2907, 2905, 2903, 2902, 2870, 2765, 2756 cm-1. The CH2 bending vibrations are presented at 1461, 1436, 1431, 1422, 1418, 1412 cm-1. The out of-plane vibrations are also identified and the values are presented in the Table 2. The values of CH2 vibrations are good agreement with the experimental data and literature [15].
N-H vibrations: Hetero cyclic compounds containing an N-H group exhibit N-H stretching absorption in the region from 3500-3200 cm-1 [20]. Hence, the N-H stretching vibrations of the title compound identified at 3350 cm-1. The in-plane bending vibrations of HCN, HNC, are observed at 1291, 1415 cm-1. The out-of-plane bending vibrations are also calculated and tabulated in Table 2.
Ring vibrations
With the help of theoretical calculations, C-N, C-C vibrations are identified and assigned in this study. The heterocyclic compound Pyrimidine’s absorb strongly at 1600-1500 cm-1 due to the C=C and C=N ring stretching vibrations. Absorptions are also observed at 1640-1620 cm-1, 1580-1520 cm-1 1000-960 cm-1 and 875-775 cm-1 [21]. Hence, the title compound have the C-N stretching modes are calculated at 1043, 1094, 1094, 1130, 1140 cm-1. The calculated C-C stretching vibrations are predicted at 967,1191,1335 cm-1. Due to the electron with drawing effect of nitrogen in the ring and oxygen in ethanol, the force constant of C-C bond is decreased and so the stretching frequency is lowered. The in-plane vibrations of HCC, CCN, HCN, CNC, HNC are identified in the range 1158-1415 cm-1 and the other values are tabulated in Table 2.
C-O and O-H vibrations: The C-O stretching vibrations in alcohols and phenols produce a strong band near 1260-1000 cm-1 and sensitive to the nature of the substituent’s bonded to carbonyl carbon. For ethyl alcohol, the intense broad band near 3360 cm-1 represents the hydrogen bonded O-H stretching vibration and the weak intensity bands at 1330 and 1270 cm-1 exhibit O-H in plane bending and C-O stretching coupled vibrations. The broad absorption near 650 cm-1 is consistent with O-H out-of-plane bending vibration [22,23].
Hence the title compound, observed the calculated value of C-O stretching at 1014 cm-1. The O-H stretching frequency is predicted at 3368 cm-1 and in plane bending vibrations of COH, HOC, COH predicted at 1170,1305, 1386 cm-1 respectively. The C-O and O-H frequencies are good agreement with available data.
13C NMR and 1H NMR analysis: The isotropic chemical shifts are frequently used as an aid in identification of reactive organic as well as ionic species. It is recognized that accurate predictions of molecular geometries are essential for reliable calculations of magnetic properties. Gauge-independent atomic orbital (GIAO) 1H and 13C chemical shift values (with respect to TMS) were calculated using the DFT (B3LYP) method with 6-31+G(d,p) basis set and compared with experimental 1H and 13C chemical shift values. The results of this calculation are shown in Table 3 together with the experimental values. The result shows that the range of 1H and 13C NMR chemical shift of the typical organic molecule is usually >100 ppm [24,25] the accuracy ensures reliable interpretation of spectroscopic parameters.
| Atoms | Experimental | B3LYP/6-31+G(d,p) |
|---|---|---|
| C16 | 58.04 | 51.00 |
| C19 | 60.91 | 47.1 |
| C7 | 54.54 | 45.24 |
| C4 | 54.54 | 45.24 |
| C10 | 45.83 | 39.87 |
| C1 | 45.83 | 39.87 |
| H20 | 3.63 | 4.007 |
| H21 | 3.63 | 4.007 |
| H17 | 2.52 | 3.203 |
| H18 | 2.52 | 3.203 |
| H3 | 2.89 | 3.108 |
| H11 | 2.89 | 3.108 |
| H12 | 2.89 | 2.87 |
| H2 | 2.89 | 2.87 |
| H5 | 2.48 | 2.74 |
| H9 | 2.48 | 2.74 |
| H8 | 2.48 | 2.48 |
| H6 | 2.48 | 2.48 |
| H14 | - | 0.554 |
| H23 | - | 0.47 |
Table 3: Experimental and calculated 13C NMR and 1H NMR chemical shift (ppm) 2-(1-piperzinyl) ethanol.
The important aspect is that, hydrogen attached or nearby electron withdrawing atom or group can decrease the shielding and move the resonance of attached proton towards to a higher chemical shift. By contrast electron donating atom or group increases the shielding and moves the resonance towards to a lower chemical shift. In the present investigation, the chemical shift values calculated for hydrogen atom (with respect to TMS) are 4.007 to 0.47 ppm. Due to the electron withdrawing atom of N13 and O22 which decreases the chemical shift value for the hydrogen atom H14 and H23. The calculated 13C chemical shift values are ranging from (with respect to TMS) 51 to 39.87 ppm. Most of the 13C and 1H chemical shift results match with the experimental data.
Frontier molecular orbitals
The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular (LUMO)are named as frontier molecular orbitals (FMO). The FMOs plays an important role in the optical and electrical properties, as well as in quantum chemistry [26]. The HOMO represents the ability to donate an electron, LUMO as an electron acceptor represents the ability to obtain an electron. The energy gap between HOMO and LUMO determines the kinetic stability, chemical reactivity and optical polarizability and chemical hardness-softness of the molecule [27-31]. In order to evaluate the energetic behavior of the title compound, the HOMO and LUMO energy gap was calculated at the B3LYP/6-31+G(d,p) level, which reveals that the energy gap reflects the chemical activity of the molecule. The energies of four important molecular orbitals of the title compound in gas: the second highest and highest occupied MO’s (HOMO and HOMO-1), the lowest and the second lowest unoccupied MO’s (LUMO and LUMO+1) were calculated. The calculated energy value of HOMO is -5.7044 eV and LUMO is -0.2019 eV in gaseous phase. The value of energy separation between the HOMO and LUMO is 5.5022 eV explains the eventual charge transfer interaction within the molecule, which influences the chemical reactivity of the molecule. The calculated HOMO and LUMO energy values are presented in the Table 4. Consequently, the lowering of the HOMO- LUMO band gap is essentially a consequence of the large stabilization of the LUMO due to strong electron-acceptor ability of the electron group. Other quantum descriptors like electronegativity (χ), chemical hardness (η), electrophilicity (ψ) and softness (ζ) of 2-(1-piperazinyl)ethanol are 5.6035, -5.8053, -0.9652, 0.8612 eV respectively, for the title molecule. The HOMO and LUMO energy diagram is shown in the Figure 3.
| Parameters | B3LYP/6-31+G(d,p) |
|---|---|
| HOMO | -5.7044 |
| LUMO | -0.2019 |
| HOMO-1 | -6.3703 |
| LUMO+1 | -0.1801 |
| HOMO-LUMO | 5.5022 |
Table 4: HOMO-LUMO energy calculated by B3LYP/6-31+G (d,p) for 2-(1-piperzinyl)ethanol.

Figure 3: HOMO and LUMO plot of 2-(1-piperazinyl) ethanol.
Analysis of molecular electrostatic potential (MESP), Mulliken atomic charges
The molecular electrostatic potential surface (MESP) is a method of mapping electrostatic potential onto the iso-electron density surface simultaneously displays electrostatic potential (electron+nuclei) distribution, molecular shape, size and dipole moments of the molecule and it provides a visual method to understand the relative polarity. Electrostatic potential maps illustrate the charge distributions of molecules three dimensionally. These maps allow us to visualize variably charged regions of a molecule. Knowledge of the charge distributions can be used to determine how molecules interact with one another. One of the purposes of finding the electrostatic potential is to find the reactive site of a molecule. In the electrostatic potential map, the semi-spherical blue shapes that emerge from the edges of the above electrostatic potential map are hydrogen atoms [31]. The molecular electrostatic potential (MEP) at a point r in the space around a molecule (in atomic units) can be expressed as
 (1)
(1)
Where, ZA is the charge on nucleus A, is the electronic density function for the molecule. The first and second terms represent the contributions to the potential due to nuclei and electrons, respectively. V(r) is the resultant at each point r, which is the net electrostatic effect produced at the point r by both the electrons and nuclei of the molecule. The total electron density and MESP surfaces of the molecules under investigation are constructed by using B3LYP/6- 31+G(d,p) method. The electrostatic potential increases in the order red

Figure 4: Molecular electrostatic potential of 2-(1-piperazinyl) ethanol.
The Mulliken atomic charge calculation has an impotent role in the application of quantum chemical calculations to molecular system [28,29] because of atomic charges effect dipole moment, molecular polarizability, electronic structure and more a lot of properties of molecular system. The charge distribution over the atoms suggests the formation of donor and acceptor pairs involving the charge transfer in the molecule. The Mulliken population analysis in 2-(1-piperaziniyl) ethanol was calculated using B3LYP level6-31+G (d,p) basis set and are listed in Table 5. All the carbon atoms are having the negative value due to the attachment of the nitrogen and oxygen atoms (N13, N15 and O22). The hydrogen atom H14 and H23 near to N13 and O22 atoms accommodate higher positive charge than the other hydrogen atoms. This is due to presence of electronegative nitrogen and oxygen atoms, the hydrogen atom attract the positive charge from the nitrogen and oxygen atoms.
| Atoms | Mulliken charges | Atomic charges |
|---|---|---|
| C1 | -0.289 | -0.29055 |
| H2 | 0.132 | 0.24053 |
| H3 | 0.152 | 0.23699 |
| C4 | -0.162 | -0.27939 |
| H5 | 0.138 | 0.24457 |
| H6 | 0.120 | 0.19444 |
| C7 | -0.162 | -0.27939 |
| H8 | 0.120 | 0.19444 |
| H9 | 0.130 | 0.24457 |
| C10 | -0.289 | -0.29055 |
| H11 | 0.152 | 0.23700 |
| H12 | 0.132 | 0.24053 |
| N13 | -0.426 | -0.71303 |
| H14 | 0.290 | 0.38740 |
| N15 | -0.124 | -0.55738 |
| C16 | -0.251 | -0.28235 |
| H17 | 0.147 | 0.24603 |
| H18 | 0.148 | 0.24603 |
| C19 | -0.017 | -0.13711 |
| H20 | 0.124 | 0.20036 |
| H21 | 0.124 | 0.20036 |
| O22 | -0.528 | -0.78573 |
| H23 | 0.346 | 0.50218 |
Table 5: Mulliken atomic charge and natural atomic charges of 2-(1-piperazinyl) ethanol.
NBO analysis
The natural bond orbital (NBO) calculations were performed using NBO 3.1 program [23] as implemented in the Gaussian 09 package at the DFT/B3LYP level in order to understand various second-order interactions between the filled orbital of one subsystem and vacant orbital of another subsystem, which is a measure of the intermolecular delocalization or hyperconjucation. A useful aspect of the NBO method is that it gives information about interactions of the both filled and virtual orbital spaces that could enhance the analysis of intra molecular interactions.
The second order Fock-matrix was carried out to evaluate the donor –acceptor interactions in the NBO basis. The interactions result in a loss of occupancy from the localized NBO of the idealized Lewis structure into an empty non-Lewis orbital. For each donor (i) and acceptor (j) the stabilization energy (E2) associated with the delocalization i→j is determined as qi donor orbital occupancy, Ei, Ej, diagonal elements, Fij, the off diagonal NBO Fock Matrix element.
 (2)
(2)
In NBO analysis large E (2) value shows the intensive interaction between electron donor and electron acceptors and greater the extent of conjugation of the whole system, the possible intensive interactions are given in the Table 3. The second order perturbation theory analysis of Fock matrix in NBO basis shows strong intra-molecular hyper conjugative interactions of σ electrons.
The strong intra-molecular hyper conjugative interaction of C1-C4 and C7-C10 from of N13, C4-H6, C7-H8 and C16-C19 from of N15 and also C19-H20 C19-H21 from of O22 of which increases ED that weakens the respective bonds leading to the stabilization of ~7.81, 9.28 and 5.74 kcal mol-1 respectively.
UV-spectral analysis
All the structures allows strong π→π* or σ→σ* transition in the UVvisible region with high extinction coefficients. TD-DFT/B3LYP/6- 31+G(d,p) calculations have been used to determine the low-lying excited states of 2-(1-piperazinyl) ethanol. The calculated results involving the vertical excitation energies, oscillator strength (f) and wavelength are carried out and it can be seen in Table 6. The calculated absorption maxima values for 2-(1-piperazinylethanol) have been found to be 255.84, 242.88 and 232.16 nm. The oscillator strength for 232.16 is higher in magnitude. These three absorption bands are mainly derived from the contribution of bands due to π→π* transitions. The theoretical spectrum of UV-VIS is shown in Figure 5.
| TD DFT/B3LYP/6-31+G(d,p) | ||
|---|---|---|
| Wave length λ (nm) | Excitation energy (eV) | Oscillator strength (f) |
| 232.16 | 5.3406 | 0.0225 |
| 242.88 | 5.1048 | 0.0094 |
| 255.84 | 4.8461 | 0.0164 |
Table 6: Theoretical electronic absorption spectra value of 2-(1-piperazinyl) ethanol.

Figure 5: Theoretically calculated UV-vis spectrum of 2-(1-piperazinyl) ethanol.
Polarizability and first order hyperpolarizability
In order to investigate the relationships among molecular structures and nonlinear optical properties (NLO), the polarizibilities and first order hyperpolarizibilities of the 2-(1-piperazinylethanol) compound was calculated using DFT/B3LYP method with 6-31+G (d,p) basis set, based on the finite field approach
The polarizability and hyper polarizability tensors (αxx, αxy, αyy, αxz, αyz, αzz and βxxx, βxxy, βxyy, βyyy, βxxz, βxyz, βyyz, βxzz, βyzz, βzzz) can be obtained by a frequency job output file of Gaussian. The mean polarizability (αtot), anisotropy of polarizability (Δα) and the average value of the first order hyperpolarizibilities (βtot) can be calculated using the equations.
αtot =α xx+αyy +αzz/3 (3)
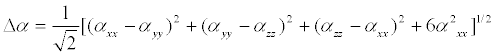 (4)
(4)
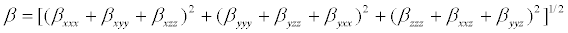 (5)
(5)
In Table 7, the calculated parameters described above and electronic dipole moment μi(i=x,y,z) and the total dipole moment μ for the title compound are listed. The total dipole moment can be calculated using the following equation.
 (6)
(6)
It is well known that the higher values of dipole moment, molecular polarizibility and first order hyper polarizability are important for more active NLO properties. The first order hyper polarizability (βtot) and the component of hyper polarizability βx, βy, βz of 2-(1-piperazinylethanol) along with the related properties (μ, αtot, Δα) are reported in Table 7. The calculated value of dipole moment was found to be 1.3549 Debye. The highest value of dipole moment is observed at 0.5692 Debye for component μx and the lowest value of the dipole moment is observed at -0.7781 Debye for component μz. The calculated polarizibility and anisotropy of polarizability 2-(1-piperazinylethanol) are 13.6492 × 10-24 e.s.u and 26.056 × 10-24 e.s.u., respectively. The magnitude of the molecular hyperpolarizibility (β) is one of the key factors in a NLO system. The B3LYP/6-31+G(d,p) calculated first order hyper polarizability (βtot) value of 2-(1-piperazinylethanol) is equal to 1.499 × 10-30 e.s.u, which is 11.5 times that of urea (0.13 × 10-30 e.s.u) [32]. This result clearly indicates that the title compound is a strong candidate to develop a nonlinear optical material.
| Parameters | Values |
|---|---|
| µx | 0.5692 |
| µy | -0.3034 |
| µz | -0.7781 |
| µtot | 1.3549 |
| αxx | 101.2163 |
| αxy | -1.4535 |
| αyy | 88.8266 |
| αxz | 8.2137 |
| αyz | -2.6602 |
| αzz | 86.2593 |
| αtot | 13.49×10-24e.s.u |
| Δα | 3240.33 |
| βxxx | 42.1762 |
| βxxy | -9.8546 |
| βxyy | 4.8639 |
| βyyy | -31.615 |
| βxxz | -12.9023 |
| βxyz | -8.7636 |
| βyyz | -41.5162 |
| βxzz | 4.4543 |
| βyzz | -4.1116 |
| βzzz | -105.0195 |
| βtot | 259.79×10-30 e.s.u |
Table 7: Electric dipole moment μ (debye), mean polarizibilityαtot (× 1024 e.s.u), anisotropy polarizibility βtot(× 1030 e.s.u) for 2-(1-piperazinyl) ethanol.
The geometrical parameters of the optimized structure were studied in detail and the influence of the substituted groups is explained. The vibrational frequency analysis by B3LYP method agrees satisfactorily with experimental results. On the basis of calculated potential energy distribution result, assignments of the fundamental vibrational frequencies have been made unambiguously. The HOMOLUMO energy gap and other related molecular properties were discussed and reported. The value of energy gap indicates that it is a good chemical reactive molecule. NBO study reveals that the lone pair orbital participates in electron donation to stabilize the compound. The MEP study shows that the electrophilic attack takes place at the N13 and O22 position of the title compound. The Mulliken atomic charges and natural atomic charges obtained are tabulated that gives the proper understanding of the atomic theory. Thus the present investigation provides complete vibrational assignments, structural information properties of the compound, 1H and 13C NMR chemical shifts values satisfactorily coincide with experimental results. The UV data indicated that the electronic transition in the compound has π-π* transitions. The values of dipole moment (μtot), linear polarizability (αtot) and first-order hyperpolarizability (βtot) of the molecule were calculated. It has been found that the value of first-order hyperpolarizability is 11.5 times greater than that of urea, which shows that the molecule is a good NLO material.