Enzyme Engineering
Open Access
ISSN: 2329-6674
ISSN: 2329-6674
Research Article - (2024)Volume 13, Issue 1
L-methioninase has significant application as chemotherapeutic anticancer drug in pharmaceutical sector. The catalytic activity of L-methioninase has depleted the exogenous supply of L-methionine by arresting growth of methionine dependent cancer cells. In this study, L-methioninase was purified from fermented broth of new bacterial isolate Pseudomonas stutzeri MTCC 101 with molecular weight (45 kDa), Michaelis constant (Km=36.5 mM) and maximum activity (Vmax=500 μmole/min/mg protein). This enzyme was resolved into 291 peptide sequences after MALDI-TOF/MS spectroscopy. Molecular docking and simulation studies as in silico approach had confirmed its higher binding affinity towards L-threonine and L-methionine substrates. The substrate specificity test had confirmed its maximum activity for specific amino acid L-methionine substrate. The high conformational stability of its three dimensional model with L-methionine substrate was validated by performing in vitro analysis against cancer cell lines. The experiments were validated by performing in vitro analysis of this enzyme against growth of cancer cell lines. This purified enzyme showed good inhibitory effect against Hepatocellular cancer (Hep-G2) and human lung carcinoma (A549) with IC50 value as 56 μg/ml and 53 μg/ml respectively. This enzyme could be could be effectively used for formulation of therapeutic anticancer drug in future.
L-methioninase; Molecular dynamic and docking; Anticancer activity; In silico; In vitro analysis
In recent year, modern immunotherapy drugs have caused subtle variations in the genetic composition of cancer patient. FDA has recently approved target specific therapy based on monoclonal antibodies and small molecules inhibitors as effective therapy for cancer patients. From last few years, some microbial amidohydrolases like asparaginase glutaminase are reported to exhibit anticancer property and received considerable attention as target specific cancer therapy [1-4]. Microbes are preferred for production of L-methioninase due to involvement of low cost purification strategy. The fermentative production of methioinase has been reported from Aspergillius flavipes, Pseudomonas putida, Brevibacterium linens, Clostridium sporogenes, Streptomyces sp. [5-8]. L-methioninase from some microbial sources has showed effective inhibitory effect on growth of colon, kidney, breast, lung and glioblastoma cancer cell lines [9,10]. L-methionine regulate cellular metabolism by synthesizing polyamines and glutathione essential for gene expression and protein biosynthesis [11]. This enzyme has been effectively formulated as anticancer drug for gastric cancer patients [12,13]. Yoshida Sarcoma and human lung tumor with minimal toxicity has showed inhibitory effect in nude mice of L-methioninase administrated drug [14].
From last few decades, the advancement in the bioinformatics tools has improved in silico analysis of binding of specific ligand for drug target therapy. The peptide sequences are highly conserved for crystal structure of protein, which helps in determining functional structure relationship of ligand with three dimensional predicted structure by performing molecular dynamic simulation process using clinical bioinformatics tools [15,16]. The presence of conserved amino acid residues within 3D structure of enzyme will enable scientists to design a strategy for improving their binding affinity for specific target ligand [17,18]. The high resolution of 3 dimensional structure of protein has been analyzed by NMR spectroscopy, X-ray crystallography and various computational methodologies. Clustal W bioinformatics tools are the most common tools used for homology modeling of query sequences against NCBI database of previously annotated sequences [19]. This approach is applicable for detection of homologous proteins exhibiting similarity of sequences only at significant number. Web tools like SWISS workspace, UCSF chimera, SWISS PDB Viewer and ExPASY’s ProtParam are employed for functional and structural characterization of predicted structure of target protein. Docking analysis has been performed for identifying the interaction of enzyme with ligand substrate and Molecular Dynamics (MD) simulation approach has further employed for analysis of stability of enzyme substrate complex in dynamic environment. There is no scientific report available for in silico analysis by molecular dynamic simulation and in vitro analysis of L-methioninase for their anticancer property till date.
In this study, biochemical characterization has been carried out for L-methioninase purified from new bacterial isolate Pseudomans stutzeri. Molecular dynamic simulation process has been done for structural and functional relationship of ligand L-methionine substrate with its predicted 3D structure. The experimental data has been further validated by performing in vitro anticancer study against different cancer cell lines.
Microorganism and culture condition
Pseudomonas stutzeri (MTCC 101) was procured from Microbial Type Culture Collection (MTCC), IMTECH, Chandigarh, India. The bacterial culture was grown on minimal media (pH 7.0) supplemented with beef extract (1 g/l), NaCl (5 g/l), yeast extract (2 g/l), peptone (5 g/l) and agar (2 g/l) at 100 rpm and 37°C in orbital shaker for 24 hours. Fermentation process for L-methioninase was carried out by adding 1% inoculums into production medium (pH 7.5 with 0.5% glucose, 0.02% malt extract, 0.1% NaCl, 0.1% casein enzymic hydrolysate and 0.025%L-methionine) and incubated flasks at 37°C for 18 hours-20 hours in an aerobic condition at 110 rpm. Fermentation broth media was centrifuged at 10,000 rpm (4°C ± 1°C) for 30 minutes and precipitate was collected simultaneously [20]. The supernatant was collected in other fresh tubes and used for the estimation of extracellular activity of L-methioninase. The pellet was re-suspended in potassium phosphate buffer (0.5 M, pH 7.0) for estimation of intracellular activity of L-methioninase. Pellet was first disrupted in ultra sonicator for 3 times for 3 minutes over 15 minutes at 20 Kc/sec and at temperature 6°C. This cell suspension was kept in alcohol: Ice mixture throughout the sonic disruptions. The treated cells were harvested at 5000 revolutions per minute for 30 minutes and extracted solutions were used for the estimation of intracellular activity of L-methioninase [21].
Assay of L-methioninase and protein estimation
The activity of L-methioninase was estimated by Nesslerization method with some modification [22]. In this method, sample containing 1% L-methionine (0.5 M phosphate buffer, pH-7.0), enzyme solution (0.5 ml), 100 mM pyridoxal-5’-phosphate was incubated at 37°C for 1 hours. The reaction condition was stopped by adding 1.5 N Trichloro Acetic Acid (TCA) and solution was centrifuged at 5,000 rpm for 5 minutes. The solution containing 0.1 ml supernatant, 3.7 ml distill water and 0.2 ml Nessler’s reagent was incubated for 20 minutes at room temperature and absorbance was noted at 480 nm. The concentration of total protein was estimate by using standard methods suggested by Lowry, et al. and Bradford, et al. [23,24].
Purification and characterization of L-methioninase
L-methioninase was partially purified at 30%-60% saturation of ammonium sulfate and pellet was re-dissolved in potassium buffer (0.05 M, pH 7.0). Enzyme was then purified by passing through Diethyl Amino Ethyl (DEAE) cellulose column (pre equilibrated with 0.05 M, pH 7.0 potassium phosphate buffer). With the same buffer column was washed and bound protein was eluted out with NaCl (0.1 M-2 M) gradient of salt. All fractions were collected and used for estimating L-methioninase activity and total protein content. The fractions showing higher activity of enzyme were pooled and their molecular weight was estimated by performing Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) [25]. On the purified enzyme, effect of pH was studied by using sodium acetate buffer pH (3-6); phosphate buffer pH (7-9); glycine NaOH buffer pH (10-12). The optimum value of pH was determined for the buffer showing maximum activity of L-methioninase after incubating enzyme mixture with different buffers. At different pH, stability of enzyme was determined by pre incubating the enzyme solution at various pH values ranging from 5.0 to 9.0 for different time interval at 4°C. On L-methioninase activity effect of temperature was measured by incubating purified samples at various temperatures (4°C to 90°C) in potassium phosphate buffer. The thermal stability of this enzyme was estimated by pre-incubating the purified enzyme in potassium phosphate buffer (pH 7.5) at different temperature (25°C, 30°C, 35°C, 37°C, 40°C, 50°C, 60°C) for different times (10 minutes-100 minutes) without substrate (L-methionine). Aliquots were removed, enzyme solution was cooled and by using standard assay method residual activity of L-methioninase was determined.
Michaelis-Menten constant (Km) and Maximum Velocity (Vmax) (Kinetic parameters) of L-methioninase (purified) were determined by Lineweaver-Burk plot by incubating enzyme with each substrate at a different concentration of (10 mM-100 mM) under optimum conditions. The turn over number (Kcat) of this enzyme was calculated using following formula:
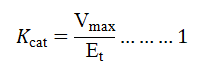
Where,
Vmax as maximum velocity and Et as total enzyme concentration
The catalytic efficiency of the enzyme (purified) was calculated as the ratio of turn over number (Kcat) and Michaeli’s constant value (Km).
L-methioninase sequencing and protein modeling
The peptide sequence for purified extract of L-methioninase from Pseudomonas stutzeri MTCC 101 was performed using MALDI-TOF/TOF mass spectroscopy. The thin gel band of SDS-PAGE containing pure extract of L-methioninase was cut, washed, de-stained in dehydrated CH3CN and dried in vacuum chamber. On ice, pieces of gel were cooled and in buffer soaked for overnight at 37°C. The mixtures were sonicated in water bath for 10 minutes for recover of digested peptides and for three times process was repeated. The tryptic digested sample after treatment with saturated solution of α-cyano-4-hydroxy-cinnamic acid (0.1% TFA and 50% acetonitrile) was dried at room temperature and used for analysis of MALDI. The protein databases (MASCOT search) for query protein samples were identified after comparing with the databases of NCBI for known proteins [26,27]. The peptide sequences for query sample were retrieved by comparing with submitted peptide sequences from different strains of Pseudomonas sp. using BLAST as a tool provided by NCBI.
X-ray crystal structure of L-methioninase from Pseudomonas putida was used as template model for prediction of tertiary structure for query peptide sequences of purified L-methioninase as shown in Figure 4. Modeller tool was employed for getting hypothetical configuration for query sequences and predicted structure was validated on the basis of Ramachandran plot analysis SWISS MODEL was used for verifying stereo chemical quality and predicted structure was analyzed in terms of psi (ψ) torsion angles for finding possibility of refinement and phi (á´?) angles for crystallographic model construction [28,29]. L-methionine (C5H11NO2S) as ligand molecule was retrieved from Zinc database and interaction energy for ligand-model structure was minimized using Merk Molecular Force Field (MMFF) [30].
Molecular docking and dynamics simulation of L-methioninase against amino acids
EA Dock DSS software was used for virtual screening, post-screening analysis and docking of predicted model structure of L-methioninase with suitable ligand substrate. In this analysis, the predicted structure of L-methioninase was docked at the binding sites of different amino acid containing ligand substrate as receptor structures using Lig Prep modules ate set physiological pH as 7.0. The default glide was combined with Induced Fit Docking protocol (IFD) for predicting interactions between model structure and substrate ligand molecules at their active site. The docking analysis through IFD has been used as an accurate method for ligand and receptor flexibility approaches [31].
Molecular Dynamic (MD) and simulations were performed for docked complex in Linux environment, for understanding conformational changes in the protein folding. In this case, Schrodinger software was used for analyzing contribution of different secondary structure within predicted model and study of association of ligand-protein structure, LINCS and SETTLE algorithms were applied for predicting the geometry and bond length of water molecules. Schrodinger tool also helped for the exploration of binding of different amino acid residues with predicted model structure as similar study performed using Xm grace tools [32-34].
In vitro anti-proliferative assessment over cancer cell lines
The cell culture for human lung carcinoma (A549) and Hepatocellular cancer (Hep-G2) were procured from the National Centre for Cell Science (NCCS), Pune, India. To various concentration viability of cell lines were exposed for determining the effect of anticancer activity on purified L-methioninase. Inhibitory Concentration (IC50) expresses anticancer activity by median growth. Dulbecco modified eagle medium supplemented with FBS (10%), penicillin-streptomycin (1%) was used for sub-culturing the cells and in CO2 incubator with CO2 (5%) incubated. Cells were counted and the viability of cells was tested after staining with trypan blue using hemocytometer. For performing MTT assay by the method cells were seeded at 1 × 104 in a 96 well plate [35].
Different cell lines were treated with different concentration of purified sample of L-methioninase and incubated (24 hours) in CO2 incubator. After 24 hours, MTT reagent (20 μl) was added and incubated (4 hours) at 37°C. Formazan was produced after incubation, and it was then solubilized by the addition of DMSO (100 μl) and by using ELISA reader absorbance was noted at 570 nm. Positive control was used for the cells without treatment with purified extract of L-methioninase. The effective dose of purified enzyme sample was estimated in terms of IC50 for inhibiting growth of 50% cancer cell.
Pseudomonas stutzeri (MTCC 101) was reported as new bacterial isolate for production of anticancer L-methioninase enzyme. The enzyme was further purified by DEAE cellulose chromatography up to 2.34 folds with specific activity of 210.4 U/mg and data for purification steps were shown in Table 1.
| Purification steps | Total activity (U) | Total protein (mg) | Specific activity (U/mg) | Purification fold |
|---|---|---|---|---|
| Crude | 296.12 | 5.38 | 55.04 | 1 |
| Ammonium sulfate (30%-60%) | 219.84 | 2.45 | 89.73 | 1.63 |
| DEAE Cellulose | 189.36 | 0.9 | 210.4 | 2.34 |
Table 1: Purification chart of L-methioninase from Pseudomonas stutzeri.
The purification of enzyme was confirmed after getting single band on casted gel plate and its molecular weight was found as 45 kDa and results were shown in Figure 1. The effect of pH on activity of L-methioninase activity was estimated by varying pH values from 3.0 to 12 and data were represented in Figure 2a. The optimum value of pH for this purified enzyme was found as 7.5 and its activity start to decrease slightly with increased value of pH from 8.0 pH values-10 pH values. The graph for pH stability Figure 2b showed its stability up to pH 5.0 to 8.0 for 12 to 14 hours at 4°C. The purified enzyme retained its 50% activity at pH 6.0 whereas; it retained 90% activity at pH 8.0. Temperature also played a significant role for regulation of enzyme structure and this enzyme showed optimum activity at 37°C as shown in Figure 2c. This enzyme showed stability up to 90 minutes at 37°C and its activity was found to be decreased slowly after incubation for 1 hour at this temperature as shown in Figure 2d.

Figure 1: Protein band of standard protein ladder (Lane 1); crude extract (Lane 2), elution extract (Lane 3), purified extract (Lane 4), partially purified extract (Lane 5) after SDS-PAGE electrophoresis.

Figure 2: Effect of environmental condition on activity of purified L-methioninase in terms of (a) Different values of pH (b) pH stability (c) Different temperature (d) Temperature stability.
The specificity of this enzyme was evaluated against different L-amino acids (L-aspartic acid, L-glutamic acid, L-alanine, glycine, L-cysteine, L-methionine, L-lysine, L-threonine and L-tyrosine) by treating with 20 mM concentration of each substrate molecule. The maximum activity was observed for L-methionine substrate and least activity was obtained with L-alanine substrate as shown in Figure 3a.
Kinetic constant values of Michaelis-Menten (Km) and maximum Velocity (Vmax) of L-methioninase (purified) were estimated by incubating enzyme with different L-methionine concentration (10 mM to 100 mM) under optimum assay conditions. The values of Km and Vmax for L-methioninase was determined by Lineweaver Burk plot (Figure 3b) as 36.5 mM and 500 μmole/min/mg protein respectively. The turnover of this enzyme was reported in term of catalytic efficiency (Kcat) value as 5 μmole/min/mg.

Figure 3: (a) Substrate specificity for effect of different amino acid substrate on the production of L-methioninase; (b) Lineweaver Burk plot for purified extract of L-methioninase.
The peptide sequences of purified L-methioninase were obtained after performing MALDI-TOF-TOF/MS spectroscopy for gel band of purified extract of L-methioninase. The enzymatic samples were hydrolyzed into 291 peptide sequences after tryptic digestion in MALDI-TOF with good signal intensity as shown in Table 2.
The query sequences are further used for prediction of 2D structure using sequences submitted in NCBI site for L-methioninase of Pseudomonas putida (Figure 4a).
| Start-End | Observed | Mr (expt) | Mr (calc) | Delta | Miss | Sequence |
|---|---|---|---|---|---|---|
| 1-17 | 1777.226 | 1776.219 | 1777.003 | -0.7844 | 1 | MRHIAIIGAGEIGQAIK.Q |
| 2-17 | 1648.132 | 1647.124 | 1645.963 | 1.1617 | 1 | M.RHIAIIGAGEIGQAIK.Q |
| 18-33 | 1964.384 | 1963.376 | 1962.966 | 0.41 | 1 | K.QLFGKQPMIFWDNDPK.K |
| 39-49 | 1233.871 | 1232.864 | 1231.677 | 1.1863 | 1 | K.KSLETVLTDAR.M |
| 59-82 | 2281.614 | 2280.606 | 2280.294 | 0.3122 | 0 | R.GLPSVLPTTAATVSTAAPIISVSK.G |
| 88-102 | 1688.171 | 1687.164 | 1685.972 | 1.1925 | 1 | K.SRLFVDELLAVSLPK.H |
| 103-121 | 2117.572 | 2116.564 | 2116.045 | 0.5197 | 0 | K.HSTALLYGPMLAEEIMANR.G |
| 122-132 | 1027.794 | 1026.787 | 1026.619 | 0.1684 | 1 | R.GAVGLAAKNK.K |
| 133-148 | 1951.324 | 1950.316 | 1950.076 | 0.2403 | 1 | K.KIYQLLHTMITVSTFR.L |
| 276-291 | 1774.247 | 1773.239 | 1772.967 | 0.272 | 0 | K.ITVSDNAPSIFQQLLK.K |
Table 2: Peptide sequence analysis of L-methioninase from MALDI-TOF/MS spectroscopy.
Ramachandran plot (Figure 4b), with 94.5% favored regions, 4.1% allowed regions and 1.4% outlier regions strongly supports the geometric fitness of the secondary structure for predicted modeled L-methioninase enzyme. The final predicted structure of the enzymatic protein was used for docking analysis for its interaction with different ligand substrate. More accurate docking images of binding of different ligand molecules like L-methionine, L-Lysine, L-Cysteine, L-Tyrosine, L-Threonine with active site of predicted 3D model structure for L-methioninase were represented in Figures 5a-5e respectively. The interpreted docking score values for these ligand substrate molecules were shown in Table 3. The superimposed images for binding of ligand substrates of L-methionine and L-Threonine were shown in Figure 6a,b. Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF), for validation of these docked model structure Schrodinger was employed. The characterization of predicted structure was further performed in terms of RMSF throughout the simulation. The composition of protein Secondary Structure Elements (SSE) like beta strands and alpha helices have been monitored. Interaction of these Protein ligand interactions have been categorized as hydrogen bonds, hydrophobic, ionic and water bridges for different ligand substrates as shown in Figure 7. The strong interaction was observed with H bond (>80%) for lysine residue (LYS-82, LYS-165) and water bridges for Asn-166 (>50%). The interaction of model structure with showed interaction of water bridges (15%) and hydrophobic interaction (5%) for Met-112 residue. The residue for Ser-81, Lys-82, Lys-165, Glu-248 also showed 10%-20% ionic bond interaction and hydrophobic interaction was solely observed for Ile-12 and Leu-113 residues. The protein ligand plot Figure 8, shows that Lys 165, Lys82, Asn 166 Gln 227 are active amino residues in predicted model structure of L-methioninase. The simulation process has summarized the conformational evolution of Rotatable Bond (RB) of model structure by the ligand torsions plot throughout the ligand simulation trajectory (0.00 through 10.00 nsec).
| S. no | Title | Entry Id | Docking score (kcal/mol) | Binding free energy (ΔG) (Kcal/mol) |
|---|---|---|---|---|
| 1 | 6288 | L-Threonine | -7.58 | -49.09 |
| 2 | 6137 | L-methionine | -6.13 | -29.77 |
| 3 | 5962 | Lysine | -4.62 | -39.41 |
| 4 | 5862 | Cysteine | -4.49 | -15.34 |
| 5 | 6057 | Tyrosine | -4.05 | -38.4 |
Table 3: Docking score chart for interaction of predicted model structure with different ligand substrate.

Figure 4: (a) Results of sequencing after BLAST analysis for query sequences for purified L-methioninase enzyme; (b) Ramachandran plot for analysis of secondary structure of predicted model for L-methioninase.

Figure 5: Docking analysis plot for predicted 3D structure of Lmethioninase with different ligand substrates (a) L-methionine (b) L-Lysine (c) L-Cysteine (d) L-Tyrosine (e) L-Threonine.

Figure 6: Superimposed images for homology modelling of predicted 3D structure of L-methioninase with template model after docking with ligand substrate (a) L-methionine (b) L-Threonine16.

Figure 7: Plot with different interaction fractions of predicted model structure with different amino acid residues by H-bond, water bridges, ionic bond and hydrophobic interaction.

Figure 8: Predicted model structure with specific L-methionine ligand substrate after docking analysis.
In vitro study was performed by adding different diluents of purified extract in cell seeded microtiter wells with Hepatocellular cancer (Hep-G2) and human lung carcinoma (A549) cell lines. This enzyme showed strong inhibitory effect on growth of cancer cell of HepG2 and A549 with IC50 value as 56 μg/ml and 53 μg/ml respectively as shown in Figure 9.

Figure 9: In vitro study of different concentration of purified L-methioninase against the growth of different cancer cell lines.
Food and Drug Administration (FDA) has recently approved target specific therapy for cancer using small monoclonal antibodies and some therapeutic enzymes. Some microbial amid hydrolase enzymes like L-asparaginase, L-methioninase, L-arginase have catalytic properties to hydrolyze exogenous source of essential amino acids required for proliferation of cancer cell. In this concern, Pseudomonas stutzeri (MTCC 101) has been explored as novel bacterial strain for producing therapeutic important L-methioninase enzyme. This enzyme was partially purified with 30%-60% saturation of ammonium sulfate which confirmed its low molecular weight range. This enzyme was effectively purified with high specific activity 210.4U/mg and 2.34 purification fold after performing DEAE-cellulose chromatography. By using SDS-PAGE, enzyme purity was analyzed for purified extract from DEAE cellulose chromatography in compare to crude extract, ammonium sulfate precipitated sample and standard protein marker. Molecular weight of purified L-methioninase was found to be 45 kDa which confirmed the low molecular weight of homogenized subunits of enzymes. Similar results are being reported for L-methioninase from Brevibacterium linens, Citrobacter freundii, Pseudomonas putida, Streptomyces sp. after purifying samples using Sephacryl S-200HR and DEAE-Cellulose column chromatography [36-39]. The low molecular weight of 35-50 kDa was found for subunits of L-methioninase from different microbial strains [40].
The purified enzyme had exhibited more activity in the range from pH 7.0 to 8.0 and its maximum activity was observed at optimum pH 7.5. L-methioninase from fungal strain like Candida tropicalis and Aspergillus flavipes showed maximum activity at 6.5-7.0 pH value [41]. Bacterial strains like Citrobacter freundii and Pseudomonas putida secreted L-methioninase with optimum pH 7.5-8.0, which was similar to our results [42]. This activity of this enzyme enhanced with increasing value of pH from 5.5 to 8.0 and decreased its activity after varying 5<pH>8.0. The decreased level of stability at lower and higher pH values might be because of denaturation of three dimensional structure of enzyme. L-methioninase from Brevibacterium linens showed stability of L-methioninase at pH ranging from 6.0 to 8.0 for 24 hours. The enzyme retained 20% activity at pH 5.5, and found to be inactivated at pH 4.0. This showed maximum activity at 37°C with 60% of relative activity for temperature varying from 30°C to 40°C and showed complete inactivation at higher temperature (above 50°C). The study of thermal stability showed that enzyme retained its maximum activity at temperature 25°C-37°C and slowly decreased at higher temperature beyond 37°C. This enzyme showed greater potential of anticancer activity due to its high thermal stability and pH stability. Many bacterial strains like Brevibacterium linens, Candida tropicalis, Citrobacter intermedius and Pseudomonas putida were also reported for exhibiting maximum activity of L-methioninase at 35°C-45°C with high thermal stability for 2 hours-4 hours [43].
The values of Michaelis-Menten (Km) and Maximum Velocity (Vmax) were estimated after treating reaction mixture with different concentration of L-methionine substrate (20 mM-100 mM) [44]. The study of steady state kinetic of enzyme catalyzed reactions predicts a hyperbolic relationship between the activity as steady state Velocity (V) and Substrate concentration (S) which is expressed in term of equation as follows:
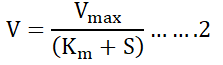
Where,
Vmax=Maximum velocity and Km=Michaeli’s constant
The low value of Km (36.5 mM) with high value of Vmax (500 μmole/min/mg) indicate higher affinity of this enzyme for L-methionine substrate which would be important study for estimation of its therapeutic values. The rectangular hyperbolic plot couldn’t be used for determining the accurate value of Vmax and Km, because of exhibiting asymptomatic factors for high values of substrate molecules [45]. The inverse values of 1/V versus 1/S in Lineweaver Burk plot were used by many biochemists for determining kinetic parameters by given equation:
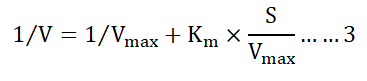
L-methioninase purified from Citrobacter freundii also showed lower Km value of 0.7 mM, whereas, Brevibacterium spp. Pinnamaneni R, et al. showed value of Km six times higher than Citrobacter intermedius. L-methioninase from Clostridium sporogenes also showed very low specificity of L-methioninase with high value of Km.
The peptide sequences obtained from MALDI-TOFF/MS spectrocopy was obtained after arranging observed tryptic peptides from N to C terminus. From NCBI database, these peptide sequences were recovered and saved in FASTA format. The crystallographic structure of L-methioninase from Pseudomonas putida was used as template model for developing predicted 3-D structure for our query peptide sequences [46]. The physicochemical properties for predicted structure were studied in terms of extinction coefficient, Isoelectric Point (pI), instability index, molecular weight by using ExPASY’s Prot-Param tools. Modeler tool was used for predicting 3D structure for query peptide sequences by multiple sequence alignment based on template peptide sequences submitted in NCBI database for Pseudomonas putida. The best predicted 3-D structure was selected on the basis of low value of RMSD and Discrete Optimized Protein Energy (DOPE) score [47,48]. The predicted secondary structure model for query sequences was analyzed using prediction protein tool with Goodness factor (G) value as 0.3. The predict structure was assumed as good quality model with acceptable range of G value varying from 0-0.5. The high value of docking score was obtained for L-threonine (-7.58) and L-methionine (-6.13) substrates. The values of positive and negative for docking score indicates the unfavorable and favorable energy environment respectively [49]. The homology modeling was performed for highly docked substrate of this predicted 3D structure using template model of L-methioninase from Pseudomonas putida. The substrate L-methionine showed high specificity for purified extract of L-methioninase than L-threonine during substrate specificity test which results were represented in histogram plot Figure 3a. Therefore, L-methionine was selected as potent ligand molecule for performing molecular dynamic and simulation process.
The study of variation of RMSD value with time showed the degree of structural conformation for docked model throughout the simulation process. The results of RMSD show the presence of small and globular proteins undergoing small conformational change during simulation process. The predicted model structure had been observed as stable at approximately 3.5 which represented its equilibrated system at the end of the simulation process. SSE distribution was reported by residue index throughout the protein structure with 39.93% helix, 11.14% strand and 48.03% other Protein Secondary Structure Elements (SSE). This model showed low degree of conformational change for beta strands and alpha helices as compared to loop regions in secondary structure elements. The interaction of different ligand substrates with the predicted model structure could be monitored throughout the simulation process. In ligand binding a significant role is played by Hydrogen bonds (H-bonds) and strong influence is caused on adsorption, metabolization and drug specificity. In this case, protein ligand H-bond showed geometric criteria with 2.5 Å distance between the (D—H•••A), donor and acceptor atoms; (H•••A—X) >90° acceptor angle between the hydrogen-acceptor-bonded atom atoms; and (D—H•••A) >120° donor angle between the donor-hydrogen-acceptor atoms. The geometric criteria for hydrophobic interactions was obtained with cation for aromatic and charged groups within 4.5 Å; for two aromatic groups stacked face-to-face or face-to-edge; and non-specific hydrophobic side chain within 3.6 Å of a ligand's aromatic or aliphatic carbons. Between two charged atoms (oppositely), ionic bond polaric interaction is observed with 3.7 Å and within 3.4 Å of ligand's heavy atoms (except carbon) and protein's metal ion is coordinated. The interaction of a protein-ligand by water molecule mediator with hydrogen bonded are defined as water bridges. With a distance of 2.8 Å, current geometric criteria for a water ligand or protein-water H-bond are observed with (D—H•••A) >110° donor angle between the donor-hydrogen-acceptor atoms, (H•••A—X) >90° acceptor angle of between the hydrogen acceptor bonded atom atoms and (D—H•••A) distance between the donor and acceptor atoms. The protein ligand plot is strongly bound to ligand substrate molecule of L-methionine after docking analysis. This ligand torsion effect shows the conformation of the strain that undergoes for maintaining ligand protein bound conformation.
The hypothetical docking analysis for the predicted model structure of this L-methioninase enzyme has attracted our research team to perform in vitro analysis of this purified enzyme extract against cancer cell. The characterization of this enzyme (purified) with maximum activity at pH 7.5, temperature 37°C and low value of Km has also strongly supported for exhibiting anticancer property. This purified enzyme showed good inhibitory effect against Hepatocellular cancer (Hep-G2) and human lung carcinoma (A549) with IC50 value as 56 μg/ml and 53 μg/ml respectively. The growth of human lung carcinoma (A549) and Hepatocellular cancer (Hep-G2) was strongly inhibited in presence of this enzyme. These results showed that L-methioninase has effectively bound to L-methionine ligand substrate within cancer cell and block the exogenous source to provide this essential amino acid and therefore showed strong inhibitory effect on its growth. These results were strongly supported by the in vitro analysis performed by other scientist for anticancer property of other L-methioninase against cell lines [50,51]. L-methioninase from Pseudomonas putida and also Hori H, et al. showed antitumoric property against some tumor cell lines [52]. L-methioninase from Aspergillus flavipes and El-Sayed AS, et al. also showed good inhibitory effect against breast cancer, liver (HEP-G2) and Prostate (PC3). Candida tropicalis also showed production of anticancer L-methioninase against breast cancer cell lines and liver cancer cell line. This purified extract of L-methioninase could be used as effective target specific therapy for lung, breast and liver cancer patients in future.
L-methioninase from Pseudomonas stutzeri MTCC 101 had significantly characterized with low molecular weight 45 kDa, and higher value of activity Vmax (500 μmole/min/mg protein). The molecular dynamic simulation process had designed three-dimensional structure for it peptide sequences and confirmed it higher binding affinity towards L-methionine substrates. In vitro analysis was further performed by treating cancer cell lines against specific doses of L-methioninase. This enzyme showed effective inhibition against growth of Hepatocellular cancer (Hep-G2) and human lung carcinoma (A549) with IC50 value as 56 μg/ml and 53 μg/ml respectively. This enzyme could be effectively used for formulation of therapeutic anticancer agent in pharmaceutical industry in future.
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Bhawana Kharayat. Priyanka Singh has contributed to the study conception, design, material preparation and analysis of experimental data. The first draft of the manuscript was written by Priyanka Singh and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
We authors declare that no funds, grants, or other support were received during the preparation of this manuscript. All authors declare that they have no relevant financial or non-financial interests to disclose.
All authors confirm that this study has not included any research work related to human subjects. The experimental data are original and have not been taken from any other’s journal article. We all authors are mutually agreed to publish this research work in this journal.
Authors are thankful to the department of Bioscience and Biotechnology, Banasthali Vidyapith (Rajasthan, India) and NIMS University (Rajasthan, India) for providing research facility to perform this work.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
Citation: Kharayat B, Singh P (2024) Therapeutic Effectiveness of L-Methioninase as Anticancer Agent: In silico Versus In vivo Analysis. Enz Eng. 13:234.
Received: 31-Aug-2023, Manuscript No. EEG-23-26412; Editor assigned: 04-Sep-2023, Pre QC No. EEG-23-26412 (PQ); Reviewed: 19-Sep-2023, QC No. EEG-23-26412; Revised: 06-Nov-2023, Manuscript No. EEG-23-26412 (R); Published: 26-Mar-2024 , DOI: 10.35248/2329-6674.24.13.234
Copyright: © 2024 Kharayat B, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.