Journal of Thermodynamics & Catalysis
Open Access
ISSN: 2157-7544
ISSN: 2157-7544
Research Article - (2016) Volume 0, Issue 0
A transition heat is the most important characteristics of first-order phase transitions. Black [1] was first who discovered in 1762 that in the transfer of water to vapor, some quantity of heat is absorbed, which he termed the latent evaporation heat. In spite of more than the two-hundred-year period of the heat transfer concept existence there are no analytical expressions relating the transition heat with other parameters of phase transitions. For example, the fundamental "Physics Encyclopedia", articles devoted to the transition heat, evaporation heat, and so on, comprises no formulae but only tables of experimental data. One can also mention monographs [2-9] which have no relationships except for the conventional definition of the transition heat λ=TΔS. Hence, obtaining the relationships between the transition heat and other parameters of first-order phase transitions will be a substantial contribution into the theory of first-order phase transitions.
Keywords: Thermodynamic; Heat; First-order phase transitions
Part I: calculation of the evaporation heat for liquid gases and metals
General expressions for the transition heat of first-order phase transitions: The conventional expression for a transition heat λ=TΔS has two substantial drawbacks. First, in some phase transitions, for example, in evaporation, not only entropy changes but a system does the work, which can only be supplied by an external source of heat. Second, the transition heat is expressed in terms of the entropy variation ΔS, which cannot be measured experimentally. In the preset work, the transition heat is defined as
λ=TΔS+A
Where T is the transition temperature in K°, ΔS is the change of system entropy, A is the work that the system does. Entropy is calculated from the general definition [3]
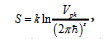
Where K is the Boltzmann factor, Vph is the volume of phase space occupied by the system, and S is the number of degrees of freedom. Thus, we obtain:
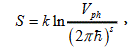
where 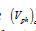 and
and 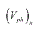 are the volumes of the old and new phases, respectively. The general expression for the heat for a phase transition has the form:
are the volumes of the old and new phases, respectively. The general expression for the heat for a phase transition has the form:
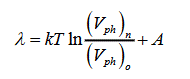 (1)
(1)
The volumes of phase spaces and the expressions for the work are specified for each particular phase transition. In the present work, all calculations are performed for one mole of substance; hence, all extensive values refer to one mole.
Approximate calculation of the phase space volumes for liquid and gaseous states
For liquid and gaseous states, the energy of system has the form:
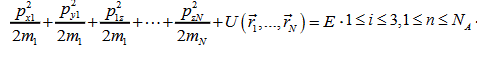 (2)
(2)
Since we consider one mole of a single-component substance, all masses are equal and N=NA is the Avogadro number. The volume of the phase space is:
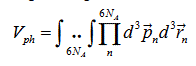
Equation (2) can be rewritten in the form:
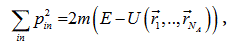
which is the equation of a 3N-dimensional sphere in 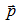 -space; hence, the 3N-dimensional integral over pulses is equal to the volume of this sphere of the radius
-space; hence, the 3N-dimensional integral over pulses is equal to the volume of this sphere of the radius 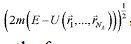 , and the expression for the volume of phase space takes the form:
, and the expression for the volume of phase space takes the form:
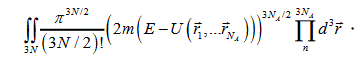
In the result of integration the dimensionality of the integral has changed from 6 to 3; however, one cannot take the rest integral without further assumptions. For calculating the 3N-dimenstional integral, let us consider the behavior of the function of kinetic energy distribution near a point of a first-order phase transition. Not specifying exactly the distribution function one may assert that it has a bell shape with a maximum that shifts with temperature to right. Near the point of phase transition such an evolution of the distribution function is impossible because the temperature of the system does not change and energy incoming still continues. Hence, the only way for the distribution function to vary is its narrowing and, in the limit, it transfers to the δ -function. In the latter case, the most probable and the average values of the kinetic energy will coincide. It is not a rigorous proof of the distribution function narrowing; however, it suggests a principal assumption of the present work, which is confirmed by a satisfactory agreement between experimental data and calculation results.
Note: Near the first-order phase transition the most of atoms (molecules, ions) are in the state with the average kinetic energy.
Since (E-U(r1,..rn)) is the kinetic energy of the system, according to the theory about equal distribution of a kinetic energy over degrees of freedom [4] one can substitute it for the average value 3NAKT/2=3RT/2, where R=KNA is the universal gas constant:
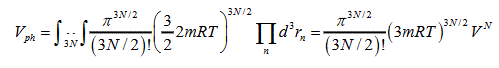
Thus, the volume of a phase space for liquid and gas is expressed as
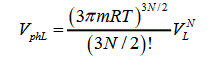 (3)
(3)
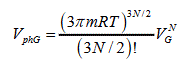 (4)
(4)
Where VL and VG are the volumes of liquid and gas, respectively.
Calculation of the evaporation heat for liquid gases
By using the general expression (1) for a transition heat one can find the expression for the evaporation heat on the saturation curve. Since the volumes of phase spaces for liquid and gas are known (3), (4), the change of entropy in evaporation has the form:
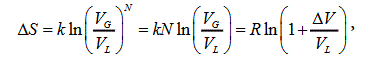
Where ΔV is the volume jump, VL is the volume of liquid, VG is the gas volume.
The work on volume expansion is A1=PΔV. In the transit liquidgas, in addition to the work on volume expansion, also the work against surface tension forces is done A2=σFN1, where σ is the surface tension coefficient, F is the area of liquid surface, N1=Va/Fd is the number of mono-molecular layers, and Va is the volume occupied by atoms (molecules, ions), d is the thickness of a mono-molecular layer (in the present work it is αr, where r is the radius of atom (molecule, ion), α=1.717 is the packing factor).
Thus, we have A2=σFN1=σFV/Fd=σVα/d.
The expression for the evaporation heat on the saturation curve has the form
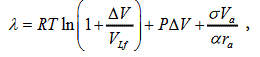 (5)
(5)
Where R is the universal gas constant; T, P are the temperature and pressure on the saturation curve, respectively; ΔV is the jump of volume in the process of evaporation; VL is the volume of liquid; Vα is the volume occupied by atoms (molecules, ions); rα is the effective atomic (molecular, ion) radius; and α=1.817 is the sphere packing factor. All extensive values refer to one mole of substance.
The expression for the evaporation heat λ comprises the volume occupied by atoms Vα and the volume occupied by liquid VL. The question arises which volume one should employ – the geometrical (experimental) volume or the free volume VLf=VLf–NAV0, where V0is the volume occupied by atom (molecule, ion). Here, the evaporation heat is calculated by using as the geometrical liquid volume VL, so and the free volume of liquid VLf.
Experimental data on the saturation curve are taken from Ref. [10], the radii of atoms and ions are taken from Ref. [11,12]. The radii of two-atomic molecules are taken as half the distance between nuclei centers [13] plus the Van der Waals radii [12]. The results are given in Table 1 and Figure 1.

Figure 1: Experimental and calculated evaporation heat versus temperature.
TK° is the evaporation temperature, P*10-5 [Pa] is the pressure, VL*105 [m3/mole] is the molar volume of liquid, ΔV*105 [m3/mole] is the jump of volume in evaporation, σ*103 [N/m] is the surface tension coefficient, r*1010 [m] is the radius of atom (molecule, ion), λex [J/mole] is the experimental value of molar evaporation heat, λT1 [J/mole] is the molar evaporation heat calculated by using the geometrical volume, λT2 [J/mole] is the molar evaporation heat by using the free volume, δ1 and δ2 [%] are inaccuracies of λT1 and λT2, respectively [14].
The calculated values of evaporation heat in Table 1 are only presented for the evaporation lines, for which the experimental values of surface tension coefficient have been found. For hydrogen, data on a surface tension coefficient along the entire evaporation curve are known; the corresponding experimental and calculated values of evaporation heat are plotted in Figure 1. A small difference between experimental and calculated values at low temperatures is related with the fact that the calculation of a phase volume should make allowance for quantum effects.
| Su-s | T | P | VL | ΔV | σ | r | λex | λT1 | δ1 | λT2 | δ2 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ne | 25 | 0.51 | 1.63 | 394 | 5.50 | 1.60 | 1790 | 1549 | 13.5 | 1757 | 1.80 |
| Ar | 90 | 1.34 | 2.90 | 533 | 10.53 | 1.92 | 6307 | 5185 | 17.8 | 5895 | 6.53 |
| Kr | 150 | 6.56 | 3.87 | 164 | 10.00 | 1.98 | 7886 | 6356 | 19.4 | 7220 | 8.44 |
| Xe | 200 | 5.22 | 4.87 | 284 | 12.00 | 2.18 | 11327 | 9100 | 19.7 | 10362 | 8.52 |
| H2 | 30 | 8.08 | 3.67 | 15.5 | 0.33 | 1.44 | 612 | 547 | 10.5 | 595 | 2.83 |
| N2 | 90 | 3.60 | 3.75 | 182 | 6.16 | 2.09 | 5057 | 3968 | 21.5 | 4668 | 7.69 |
| O2 | 100 | 2.55 | 2.95 | 303 | 10.70 | 2.00 | 6490 | 5256 | 19.0 | 6209 | 4.32 |
| F2 | 95 | 2.78 | 2.65 | 261 | 10.70 | 2.06 | 6775 | 5024 | 25.9 | 6426 | 5.16 |
| Cl2 | 201 | 0.13 | 4.26 | 14570 | 33.00 | 2.47 | 21934 | 18408 | 16.1 | 22110 | -0.80 |
| CH4 | 105 | 0.56 | 3.70 | 1521 | 15.80 | 2.30 | 8390 | 7333 | 12.6 | 8872 | -5.74 |
T K° is the evaporation temperature, P*10-5 [Pa] is the pressure, VL*105 [m3/mole] is the molar volume of liquid, ΔV*105 [m3/mole] is the jump of volume in evaporation, σ*103 [N/m] is the surface tension coefficient, r*1010 [m] is the radius of atom (molecule, ion), λex [J/mole] is the experimental value of molar evaporation heat, λT1 [J/mole] is the molar evaporation heat calculated by using the geometrical volume, λT2 [J/mole] is the molar evaporation heat by using the free volume, δ1 and δ2 [%] are inaccuracies of λT1 and λT2, respectively.
Table 1: Calculation results of evaporation heat.
From Table 1 and Figure 1 one can see that the calculations of the evaporation heat performed by the obtained formula well agree (within several percent) with experimental results. Hence, the assumption, that the most of atoms (molecules, ions) near a point of a first-order phase transition are in the state with the average kinetic energy, is valid. Also valid is the assumption that in a calculation of the evaporation heat one should take into account the work done by the system. The employment of the free volume in calculations also gives a better agreement between experimental and calculated results. Thus, one can assert that the molar evaporation heat λ on the saturation curve is described by the expression:
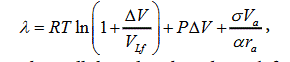 (6)
(6)
where all the values have been defined above.
Specific features of calculating the evaporation heat for liquid metals
As one can see from the results presented in Table 1, calculations of evaporation heat should be performed with the "free volume". For determining the "free volume" of liquid metals one should know the radii of ions. Handbooks [11,12] comprise two metal ion radii: radii for ions M +1, M +2 and so on, and for metal. Since it is not clear which radius should be used in the calculations of the free volume, the latter was calculated by using as the metal radius so and the ion radius. The results with the employment of the ion radius are given in Table 2, and the results based on metal radius are presented in Table 3.
| El-s | T | P | VL | ΔV | σ | ri | Vi | VLf | λex | λT | δ |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Li | 1600 | 0,91 | 1,73 | 13058 | 273 | 0,78 | 0,12 | 1,61 | 134720 | 133995 | 0,5 |
| Na | 1150 | 0,96 | 3,09 | 8760 | 120 | 0,99 | 0,24 | 2,84 | 89590 | 86899 | 3,0 |
| K | 1000 | 0,73 | 5,82 | 10516 | 65 | 1,33 | 0,59 | 5,23 | 75994 | 72581 | 4,5 |
| Rb | 950 | 0,92 | 7,25 | 7893 | 50,7 | 1,49 | 0,83 | 6,42 | 69742 | 65089 | 6,7 |
| Cs | 800 | 0,20 | 8,56 | 31264 | 47,5 | 1,69 | 1,22 | 7,34 | 65126 | 63799 | 2,0 |
| Hg | 613 | 0,75 | 1,57 | 6820 | 384 | 1,12 | 0,35 | 1,22 | 59275 | 56159 | 5,3 |
T[K] is the evaporation temperature, P*10-5 [Pa] is the pressure, VL*105 [m3/mole] is the molar volume of liquid, δV*105 [m3/mole] is the volume jump in evaporation, σ*103 [J/m2] is the surface tension coefficient, rm*1010 [m] is the ion radius in Table 2 and the metal radius in Table 3, Vm*105 [m3/mole] is the volume occupied by ions, VLf*105 [m3/mole] is the "free volume" of liquid, λex [J/mole] is experimental evaporation heat, λT [J/mole] is calculated value of evaporation heat, and δ % is the calculation error λT.
Table 2: Calculation results for the evaporation heat on the saturation curve for metals by using the ion radii.
| El-s | T | P | VL | ΔV | σ | rm | Vm | VLf | λex | λT | δ |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Li | 1600 | 0,91 | 1,73 | 13058 | 273 | 1,57 | 0,97 | 0,75 | 134720 | 151512 | -12,5 |
| Na | 1150 | 0,96 | 3,09 | 8760 | 120 | 1,89 | 1,70 | 1,39 | 89590 | 98320 | -9,7 |
| K | 1000 | 0,73 | 5,82 | 10516 | 65 | 2,36 | 3,31 | 2,51 | 75994 | 82311 | -8,3 |
| Rb | 950 | 0,92 | 7,25 | 7893 | 50,7 | 2,53 | 4,08 | 3,17 | 69742 | 73767 | -5,8 |
| Cs | 800 | 0,20 | 8,56 | 31264 | 47,5 | 2,74 | 5,18 | 3,37 | 65126 | 72209 | -10,9 |
| Hg | 613 | 0,75 | 1,57 | 6820 | 384 | 1,60 | 1,03 | 0,54 | 59275 | 67675 | -14,2 |
T[K] is the evaporation temperature, P*10-5 [Pa] is the pressure, VL*105 [m3/mole] is the molar volume of liquid, ΔV *105 [m3/mole] is the volume jump in evaporation, σ*103 [J/m2] is the surface tension coefficient, rm*1010 [m] is the ion radius in Table 2 and the metal radius in Table 3, Vm*105 [m3/mole] is the volume occupied by ions, VLf*105[m3/ mole] is the "free volume" of liquid, λex[J/mole] is experimental evaporation heat, λT[J/mole] is calculated value of evaporation heat, and % is the calculation error λT.
Table 3: Calculation results for the evaporation heat on the saturation curve for metals by using the metal radii.
T[K] is the evaporation temperature, P*10-5 [Pa] is the pressure, VL *105 [m3/mole] is the molar volume of liquid, ΔV *105 [m3/mole] is the volume jump in evaporation, σ*103 [J/m2] is the surface tension coefficient, rm*1010 [m] is the ion radius in Table 2 and the metal radius in Table 3, Vm*105 [m3/mole] is the volume occupied by ions, VLf*105 [m3/mole] is the "free volume" of liquid, λex [J/mole] is experimental evaporation heat, λT [J/mole] is calculated value of evaporation heat, and δ % is the calculation error λT.
From Table 2 one can see that the employment of ion radii in calculations of the free volumes of liquid gives a good agreement with experimental data; however, the presence of free electrons in liquid and screening of ions make one to assume that more realistic are metal radii. Results of calculations with metal radii are given in Table 3. One can see that the calculated values of the evaporation heat in this case are systematically greater than the experimental values. This can be explained by the fact that in evaporation of metals, in addition to endothermic processes, there are also exothermic processes.
Recombination of ions and electrons with the origin of neutral atoms occurs for all metals at the interface liquid-gas (vapor). In this case, the energy is released and completely or partially participates in the evaporation process. In addition, alkali metal atoms in the gaseous state form two-atomic molecules [10] with an energy release. Thus, Δλ=λT- λex is the evaporation heat received by a system due to the exothermic processes considered above. The energy released due to generation of two-atomic molecules is proportional to the fraction of two-atomic molecules in gas, and the energy released in the ion recombination is constant and independent of thermodynamic parameters.
Hence,
Δλ=βC+Q0, (7)
Where C is the part of two-atomic molecules in gas, β is the energy, released in the process of producing 0.5 mole of two-atomic molecules, Q0 is the heat released in recombination of one mole of metal ions.
Calculation results for the evaporation heat, experimental values of evaporation heat in a wide temperature range on the evaporation curve for all alkali metals and mercury, and data on the part of two-atomic molecules in gas for alkali metals are given in Table 4.
| El-s | T | P | VL | ΔV | σ | rm | Vm | VLf | λex | λT | C | Δλ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Li | 1400 1500 1600 1700 1800 |
0,18 0,43 0,91 1,77 3,19 |
1,64 1,69 1,73 1,77 1,82 |
64895 29223 14622 7983 4691 |
265,5 251,5 237,5 223,5 209,5 |
1,55 1,55 1,55 1,55 1,55 |
0,939 0,939 0,939 0,939 0,939 |
0,701 0,751 0,791 0,831 0,881 |
139563 137114 134719 132394 129487 |
154081 153172 152305 151553 150689 |
8,67 10,55 12,60 13,57 15,05 |
14518 16058 17586 19159 21202 |
| Na | 900 1000 1100 1200 1300 1400 1500 |
0,05 0,20 0,60 1,50 3,22 6,26 11,01 |
2,87 2,95 3,05 3,15 3,25 3,37 3,49 |
145285 41885 15340 6636 3360 1859 1129 |
145,3 135,3 125,3 115,3 105,3 95,3 85,3 |
1,89 1,89 1,89 1,89 1,89 1,89 1,89 |
1,702 1,702 1,702 1,702 1,702 1,702 1,702 |
1,168 1,248 1,348 1,448 1,548 1,668 1,788 |
95107 92849 90628 88481 86469 84434 82547 |
102618 102068 101145 100064 99339 98268 97294 |
6,37 8,58 10,79 12,89 14,74 16,58 18,12 |
7511 9219 10517 11583 12870 13834 14747 |
| K | 800 900 1000 1100 1200 1300 1400 |
0,06 0,24 0,73 1,86 3,91 7,30 12,44 |
5,43 5,62 5,82 6,04 6,28 6,54 6,81 |
108807 30668 11355 4904 2703 1475 899 |
78,75 72,15 65,55 58,95 52,35 45,75 39,15 |
2,36 2,36 2,36 2,36 2,36 2,36 2,36 |
3,313 3,313 3,313 3,313 3,313 3,313 3,313 |
2,117 2,307 2,507 2,727 2,967 3,227 3,497 |
79866 77942 75944 73899 71823 69739 67674 |
85079 84272 83606 82454 82814 80698 78990 |
4,05 5,98 7,97 9,88 11,61 13,10 14,31 |
5213 6330 7662 8455 10991 10959 11316 |
| Rb | 700 800 900 1000 1100 1200 1300 |
0,03 0,16 0,55 1,47 3,30 6,47 11,43 |
6,61 6,85 7,11 7,39 7,70 8,03 8,40 |
183451 42015 13667 5661 2768 1535 937 |
65,0 59,35 53,55 47,75 41,95 36,15 30,35 |
2,53 2,53 2,53 2,53 2,53 2,53 2,53 |
4,082 4,082 4,082 4,082 4,082 4,082 4,082 |
2,528 2,768 3,028 3,308 3,618 3,948 4,318 |
72632 70718 68777 66862 64931 62999 61075 |
76715 76303 75483 74680 73783 72816 71729 |
3,44 5,46 7,61 9,70 11,62 13,26 14,62 |
4083 5585 6706 7818 8852 9817 10655 |
| Cs | 700 800 900 1000 1100 1200 1300 |
0,04 0,20 0,66 1,69 3,63 6,79 11,41 |
8,26 8,56 8,89 9,24 9,65 10,1 10,7 |
132419 32798 11300 4843 2478 1440 924 |
50,3 47,5 42,7 37,9 33,1 28,3 23,5 |
2,68 2,68 2,68 2,68 2,68 2,68 2,68 |
4,851 4,851 4,851 4,851 4,851 4,851 4,851 |
3,409 3,709 4,039 4,389 4,799 5,249 5,849 |
69366 67731 66016 64209 62361 60487 58600 |
72071 71982 71321 70410 69605 68785 67779 |
3,03 4,91 6,95 8,98 10,86 12,05 13,85 |
2705 4251 5305 6201 7244 8298 9179 |
| Hg | 373 423 473 523 573 623 |
0,0004 0,0038 0,0232 0,0996 0,3302 0,8990 |
1,50 1,52 1,53 1,54 1,56 1,57 |
8285191 931431 169895 43651 14417 5748 |
452 439 429 416 402 378 |
1,55 1,55 1,55 1,55 1,55 1,55 |
0,939 0,939 0,939 0,939 0,939 0,939 |
0,561 0,581 0,591 0,601 0,621 0,631 |
60848 60518 60194 59872 59546 59206 |
70421 69241 68471 67662 66801 65695 |
9573 8723 8277 7790 7255 6489 |
T [K] is the evaporation temperature, P*10-5 [Pa] is the pressure, VL*105 [m3/mole] is the molar volume of liquid phase, ΔV*105 [m3/mole] is the volume jump, σ*103 [J/m2] is the surface tension coefficient, rm*1010 [m] is the metal radius for ion, Vm*105 [m3/mole] is the volume occupied by ions, VLf*105 [m3/mole] is the "free volume" of liquid, λex [J/ mole] is the experimental value of evaporation heat, λT [J/mole] is the calculated value of evaporation heat, C[%] is the concentration of two-atomic molecules in a gas of alkali metals, Δλ=λex-λT [J/mole].
Table 4: Calculation of Δλ.
Basing on these data, dependences Δλ=f (C) for alkali metals were plotted in Figure 2. One can see that the assumption about linear dependence of Δλ on C is confirmed with a high accuracy. Moreover, one can assert that the energies released in generating two-atomic molecules Na2, K2, Rb2, Cs2 are similar, and the energy released in generating Li2 is substantially higher. The values of Q0 are, respectively, Q0Li=4510 J/mole, Q0 Na=3510 J/mole, Q0 K=2510 J/mole, Q0 Rb=2030 J/ mole, and Q0 CS=1220 J/mole and well correlate with the ionization energy for these metals. This does not mean that the linear dependence will still exist at very high temperatures close to the critical temperature, because λex and λT tend to zero as the temperature approaches the critical value; hence, Δλ also tends to zero. The mechanism of λex reduction at high temperatures is most probably related with the fact that an exothermic process of dissociation of two-atomic molecules starts. Unfortunately, there are no experimental data for calculating λT and no information about the behavior of λex and the part of two-atomic molecules in gas at high temperatures close to the critical temperature.

Figure 2: Dependence of Δλ=λex-λT on the concentration of two-atomic molecules in gas for alkali metals.
In Figure 3 one can see a dependence of Δλ on temperature for mercury. A linear extrapolation of Δλ=f (T) turns Δλ to zero at a point close to the value of Tc.

Figure 3: Dependence of Δλ=λex-λT on temperature for mercury.
We may assert that suggested expression (6), which only comprises measurable parameters, allows one to calculate the evaporation heat of liquid gases with a high accuracy taking into account considerations of Sect. 4, and the evaporation heat of liquid metals.