Journal of Clinical Trials
Open Access
ISSN: 2167-0870
ISSN: 2167-0870
Research Article - (2016) Volume 6, Issue 2
Objectives: Before executing a clinical trial there is a lot of administrative work to do. The initiator of the study has to ask for and create several essential documents such as contracts, study protocol and case report form to name but a few. The aim of this research was to identify the current main challenges and time consumption during this planning phase of an investigator initiated trial (IIT).
Methods: A survey was conducted among monitors, principal investigators/study nurses, sponsors, study managers, federal authorities and ethics committees to capture the durations and delays of creating the Essential Documents of a clinical trial and their transmissions to the corresponding addressee. The questionnaire dealt with general information about the participant, the essential documents according to good clinical practice (GCP), the study protocol and the process of approving the documents and consequently the clinical trial.
Results: The responses showed that 95% of the participants maintain that the creation of the Essential Documents and waiting for the contributors is the most time consuming process in the planning of a clinical trial. Thereby the study protocol, contracts and (electronic) case report forms ((e) CRF) take 48% of the time in order to create all 20 Essential Documents. For that matter the survey showed that waiting for responses from reviewers and second examiners takes more than five weeks in average. Furthermore it showed that the coordination between the involved parties (45%) is a major time factor during the creation of the study protocol.
Conclusions: The survey identifies the enormous potential for optimizing the creation and transfer of the essential documents during the planning phase of a clinical trial. Furthermore it shows that the planning and coordination between editors, contributors and reviewers are a significant bottleneck before starting a clinical trial and should be supported better.
Keywords: Investigator; Survey; Protocol; Good clinical practice; Ethics
The first step is always the hardest. That applies more than ever for conducting clinical trials. The investigator has to do a lot of administrative work before conducting a clinical trial and is responsible for generating all essential documents which are elementary and “serve to demonstrate the compliance of the investigator, sponsor and monitor with the standards of Good Clinical Practice and with all applicable regulatory requirements” [1,2]. These essential documents consist of 20 documents including study information, contracts, study protocol, reports and case report form (Table 1).
| Title of Document | Purpose |
|---|---|
| Investigator’s brochure | To document that relevant and current scientific information about the investigational product has been provided to the investigator |
| Signed protocol and amendments and sample case report form (CRF) | To document investigator and sponsor agreement to the protocol/amendment(s) and CRF |
| Information given to trial subject | To document the informed consent, to document that subjects will be given appropriate written information (content and wording) to support their ability to give fully informed consent, to document that recruitment measures are appropriate and not coercive |
| Financial aspects of the trial | To document the financial agreement between the investigator/institution and the sponsor for the trial |
| Insurance statement | To document that compensation to subject(s) for trial-related injury will be available |
| Signed agreement between involved parties | To document agreements |
| Dated, documented approval/ favourable opinion of Institutional Review Board (IRB)/ Independent Ethics Committee (IEC) | To document that the trial has been subject to IRB/IEC review and given approval/ favourable opinion. To identify the version number and date of the document(s) |
| Institutional Review Board/ Independent Ethics Committee composition | To document that the IRB/IEC is constituted in agreement with GCP |
| Regulatory Authority(ies) authorisation/ approval/ notification of protocol | To document appropriate authorisation/ approval/ notification by the regulatory authority(ies) has been obtained prior to initiation of the trial in compliance with the applicable regulatory requirement(s) |
| Curriculum vitae and/ or other relevant documents evidencing qualifications of investigator(s) and sub-investigator(s) | To document qualifications and eligibility to conduct trial and/or provide medical supervision of subjects |
| Normal value(s)/ range(s) for medical/ laboratory/ technical procedure(s) and/ or test(s) included in the protocol | To document normal values and/or ranges of the tests |
| Medical/ laboratory/ technical procedures/ tests | To document competence of facility to perform required test(s) , and support reliability of results |
| Sample of label(s) attached to investigational product container(s) | To document compliance with applicable labelling regulations and appropriateness of instructions provided to the subjects |
| Instructions for handling of investigational product(s) and trial-related materials | To document instructions needed to ensure proper storage, packaging, dispensing and disposition of investigational products and trial-related materials |
| Shipping records for investigational product(s) and trial-related materials | To document shipment dates, batch numbers and method of shipment of investigational product(s) and trial-related materials. Allows tracking of product batch, review of shipping conditions, and accountability |
| Certificate(s) of analysis of investigational product(s) shipped | To document identity, purity, and strength of investigational product(s) to be used in the trial |
| Decoding procedures for blinded trials | To document how, in case of an emergency, identity of blinded investigational product can be revealed without breaking the blind for the remaining subjects’ treatment |
| Master randomisation list | To document method for randomisation of trial population |
| Pre-trial monitoring report | To document that the site is suitable for the trial |
| Trial initiation monitoring report | To document that trial procedures were reviewed with the investigator and the investigator’s trial staff |
Table 1: Essential documents needed for planning a clinical trial [1].
Additionally the current workflow entails a review phase for the involved parties. Very often a study manager (study management) is already involved in the document creation phase in order to reduce the occurrence of formal errors. The amount of time needed for the whole creation process including the review is difficult and almost impossible to identify. That is because every clinical trial is unique (unless it is a follow-up study) and the duration of planning the trial and generating the documents varies from trial to trial. Furthermore the review of the documents does not take the same time for every trial. But time slots are given for the reviewer so that at the very least an assumption with the longest possible duration is possible.
For a clinical trial however it is necessary to get an initial indication of how long the intended trial will take. Normally this assumption is based on former experiences with similar trials. But there are no reference values about the planning phase from other investigators or trials. There are a few research articles which cover the planning of an entire clinical trial [3] or for some critical events [4], but not explicitly the planning phase of a clinical trial. In addition to that both articles are quite outdated. To get a better overview of the time needed and to do a rough estimation for planning a “standard” (average) clinical trial this survey was created.
Additionally, this work shall identify the main challenges and time expenses during the current process workflow for planning a clinical trial. The aim was to capture the durations and delays of creating the Essential Documents in terms of their transmissions to the corresponding addressee.
From June 2015 until November 2015, a survey was conducted among monitors, principal investigators/study nurses, sponsors, study managers, federal authorities and ethics committees. Apart from the federal authorities and ethics committees all other participants involved in studies participated in this survey.
The survey was conducted by means of an online questionnaire and was sent to different hospitals, organizations and networks for them to forward and complete. The first part of the questionnaire dealt with general information about the participant of the survey. The second section was regarding the essential documents according to good clinical practice (GCP). The third group of questions is specialized with the study protocol and the last part of the survey is about the process of approving the documents and consequently the clinical trial.
In total 18 questions were used to gather the desired information. Different types are used for the questions and the corresponding answering options. The used types are:
• Matrix questions
• Yes/no questions
• Single/ Multiple choice with additional options
• Scaled questions
• Classification questions
• Open ended questions.
The online survey was realized with the free and open source application LimeSurvey [5]. Login to the system was not required to complete the survey. Furthermore no identifiable information was gathered and therefore the participation was fully anonymous and backtracking to the participants was not possible.
In total 38 participants from all over Germany with the combined experiences of more than 1500 conducted studies in the last five years are recorded. This shows that the people who completed this survey possess an enormous amount of expertise.
About one in two of the participants were an investigator or study nurse and one in three was part of the study management. The rest of the attendees are spread between sponsors, clinical research associates and other roles of a clinical trial.
Essential documents
As already mentioned there is a lot of documentary and administrative tasks to be done before a clinical trial can be conducted. The answers show that 95% of the participants think that creating the essential documents and waiting for the contributors is the most time consuming process in the planning of a clinical trial. Thereby the three documents, respectively document types, study protocol, contracts and (e) CRF, take 48% of the time in order to create all 20 Essential Documents. Especially the study protocol, according to the participants, takes the longest with 21% of the time until completion. Thus the preparation of the study protocol takes on average up to nine weeks whereas the time to complete all essential documents is around 15 weeks.
Time factors during the creation of the study protocol are coordination between the involved parties (45%), the waiting and idle time (35%) and the planning of the processes (20%). Additionally the survey showed that the time spent waiting for responses from reviewers and second examiners are longer than five weeks in average.
Transmission
The response from the reviewers was mostly sent by email (81.1%) and accordingly very rarely with post or “other” transmission types. The table below shows what types of transmissions is used between the author and the reviewers, sponsors, federal authorities and ethics committees (Table 2).
| Transmission | ||||
|---|---|---|---|---|
| Fax | Post | Other | ||
| To reviewer | 76.30% | 0% | 21.10% | 2.60% |
| From reviewer | 81.10% | 0% | 13.50% | 5.40% |
| To sponsor, federal authorities and ethics committees | 29.70% | 0% | 62.20% | 8.10% |
Table 2: Used transmission types during planning phase in clinical trials.
Approval respond
Sponsors, federal authorities and ethics committees have a time limitation until they respond to the author of the documents. Academic sponsors usually use their own (private) standard operating procedures (SOPs) to limit their response time. These times differ from one sponsor to the other and aren’t standardized. But also federal authorities and ethics committees are bound by deadlines. In §42 of the German Medicines Law [6] the time period of the responses is restricted to 30 to 60 days (depending on the clinical trial). Within this law there is a distinction between different types of studies whereas the conducted survey asked for the time in general with no formal or informal objections. The answers yield that waiting for the sponsor response takes about 21 days, the federal authorities response more than 42 days and the ethics committees response more than 40 days. The given time values are meant for study protocols without or with just minor objections. For protocols with major objections the time until completion increases correspondingly.
Objections from constitutions whose approval is needed can be in form or content. The frequency of occurring objections (either in form or content) until a study protocol is approved is shown in Figure 1. The results illustrate that there is a similar distribution between all three constitutions with two oppositional spikes for the ethics committees (‘Never, ‘Always’).
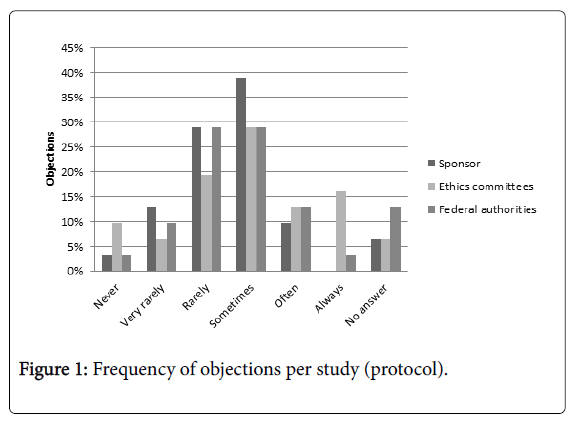
Figure 1: Frequency of objections per study (protocol).
Sponsor and ethics committees have their peaks at ‘Sometimes’ with 39% respectively 29% and federal authorities at ‘Rarely’ and ‘Sometimes’ with 29% in each case.
The outcome revealed that the creation and completion of the essential documents is a very time consuming task. To prepare all the documents with correct form and content can be very frustrating. Therefore it is suggested that investigators contact a clinical research organisation (CRO) or local study centers quite early in the process to support them while creating the documents. This assistance can lead to fewer objections and reduce the time wasted in correcting these errors.
Furthermore, it is important to improve the communication and collaboration between the involved parties which can reduce about 80% of the time-sinks during the planning phase.
This result from the fact that after the initial preparation of the documents, the waiting times on the responses of the contributors and reviewer, as well as the back and forth of the documents is remarkable. The fact that the documents transmission to federal authorities is mostly done by post is also quite alarming. This situation also explains the huge time period for the planning of clinical trials. Each participant of the process uses their own process or procedures. But with the Clinical Trials Regulation (CTR) EU No 536/2014 some changes are coming up to optimise the actual workflow. This regulation “will ensure that the rules for conducting clinical trials are identical throughout the EU” [7]. It also governs an EU-wide approval process for clinical trials. The implementation of this regulation will offer a new portal hosted by the European Medicines Agency (EMA) [8]. This portal provides functionality for an electronic submission of the intended clinical trial and will not be finished until end of May 2016.
For all that, a business process for the complete clinical trial planning process would be very valuable and can be a huge opportunity to reduce the time consumption while creating the final documents. With a standardized process time-sinks and objections can be minimized or even eradicated (at least formal objections). Furthermore it illustrates the optimization chances of the collaborative cooperation between the involved parties and consequently of the whole planning phase of a clinical trial [8-11].
I would like to thank all participants and the Coordination Centre for Clinical Trials (KKS) Duesseldorf. Special thanks to Alexandra Kley (KKS) who supported with her expertise in study management to create the survey and Henrike Kolbe (KKS) for her expertise in sponsorship and supporting by disseminating the survey.