Journal of Horticulture
Open Access
ISSN: 2376-0354
ISSN: 2376-0354
Research Article - (2014) Volume 1, Issue 3
Flowers are the most complex and attractive organ in flowering plants that play an important role in the angiosperm reproductive process. In this study, we employed next-generation high-throughput sequencing (RNA-Seq) to gain insight into the wide range of transcriptional events that are associated with petal development in the Chinese cabbage (Brassica rapa ssp. pekinensis). We isolated RNA sequences from the vegetative mass at the four-leaf stage and petals in their full-bloom. The sequences were aligned to a reference genome and analyzed to measure gene expression levels and to detect Differentially Expressed Genes (DEGs). A total of 11,079 DEGs were obtained, of which 3,900 DEGs were up-regulated in the petal sample, and 7,179 were down-regulated. In 1,595 Specifically Expressed Genes (SEGs); 303 SEGs exhibited up-regulation with 1,292 down-regulations. Gene Ontology (GO) analysis was further used to recognize the main biological functions of DEGs. Subsequently, using KEGG enrichment analysis, we found that DEGs were involved in 125 pathways. Finally, quantitative real-time PCR (qRT-PCR) was used to verify the expression pattern of partial DEGs against the sequencing data. Importantly, all partially known genes (which were specifically expressed in petals) were reflected in the attained DEGs, including; PISTILLATA, APETALA3, SEPALLATA2, SEPALLATA1. Moreover, we found multiple genes that gave high levels of expression differences but whose functions were unknown. Further functional analysis of the differentially expressed genes set will help us to elucidate the mechanism of petal development and may significantly facilitate the breeding practices at the molecular level.
<Keywords: Chinese cabbage (Brassica rapa ssp. pekinensis); Petal; RNA-seq; Transcriptome; Differentially expressed genes
Flowers are the most complex and attractive organ in flowering plants that play an important role in the angiosperm reproductive process. For thousands of years, researchers have attempted to reveal the mechanisms of flower development. Petal shape and color are the most distinctive features which allow classification of the different families of seedling plants. Furthermore, the bright colors of petals and their fragrance attract insects for pollination and can significantly affect the seed-setting rate [1]. In order to gain an insight into the molecular mechanism of petal development and its role in reproduction, we must first study its origins, development and development related genes.
To date most studies on flowering have been limited to their morphological description and physiological development. There are three physiological developments that must occur in order for this to take place: firstly, the plant must pass from sexual immaturity into a sexually mature state (i.e. a transition towards flowering); secondly, the transformation of the budding function from a vegetative mass into floral maturation; and finally the growth of the flower’s individual organs. For example, Smyth et al. [2] observed the flower development patterns of the Arabidopsis embryo from its primordial stage to flowering, by dividing the development characteristics into 20 independent stages. Their study was successful in providing the morphological basis for investigations of its molecular mechanism. In recent years, scientists have successfully discovered the heterosexual homologous mutant in both the Arabidopsis and Antirrhinum flowers, which form the basis of research for genetic diversity of derivation of floral organs [3-5]. In the research findings of these homeotic mutants, Weigel and Meyerowitz [6] have proposed the prominent ‘ABC’ model hypothesis [6-9], which endeavors to describe the biological basis of the process from the perspective of molecular and developmental genetics. In nature, flowers are colorful and have various dimensions and shape, however, the basic configuration is similar for most flowers; consisting of four different types of organs, i.e. the sepal, petal, stamen and carpel. In development of dicotyledon floral organs (containing two embryonic leaves), the basic unit is whorls, where 1-4 whorls yield sepal and carpel. When the ABC model holds, there are three class genes in the four whorls of dicotyledons floral organ. Each class genes controls the floral organ development of adjacent two whorls. For example, class A genes (including APETALA 1(AP1), APETALA 2(AP2) ) determines the sepal development, class B genes (APETALA 3(AP3), PISTILLATSA (PI)) along with class A genes determine the petal development, class B genes plus C together determines the stamen development, class C genes (AGAMOUS(AG)) alone control the carpel development. While there is antagonism between class A and C genes [10] these three ABC type genes are classed as homeotic and involved in the floral organ-specific development process of primordial floral and commonly expressed at a particular time and space, to ensure normal development of floral organs.
The aim of the current study was to focus on the petal development related with class A and B genes. The comparative study of class A, B, C genes in different species has previously shown that, class B and C genes have a strikingly conservative property [11-14]. In contrast, class A genes are less distinctive and thus difficult to determine [15]. Hence studies of petal development have predominantly focused on class B genes [16].
Nevertheless, in addition to the calyx, corolla, stamen and pistil, a structure known as the ovule exists in the floral organ. This structure gives rise to and contains the female reproductive cells. Angenent et al. [17] have found that the corresponding FB97/11 gene can control the development of petunia embryos, and subsequently the BFL1 and SIN 1 influence the development of the Arabidopsis ovule. Therefore, an extension has been proposed to modify the ABC model to the ABCD model.
In morphology, scientists have considered petals to phyllome [18], where the petals are classed as the modified leaf. Therefore, if a gene has been enabled, the leaves and petals are able to inter-change. The study demonstrates that the floral organ development status can be altered artificially by controlling the ABC genes, but does not transform the leaves into flower organs [19,20]. This illustrates that a new class of genes exist which can force the vegetative to change into floral organs. In recent studies, researchers have demonstrated this phenomena through the SEP genes of Arabidopsis, also known as the E-function genes [21,22]. In Arabidopsis the expression of ABC genes combined with SEP genes allow leaves to develop into intact floral organs, and proves that ABCE genes have the combined effects to determine the characteristics of floral organs [18,21]. Thus a quartet model has been proposed [10,23] which permits the formation of petals based on protein complexes, AP1-AP3-PI-SEP.
At present, only a few genes have been detected where molecular and genetic mechanisms have been related to petal development [24-31]. Based on these studies, several genes are capable of controlling petal growth by affecting cell proliferation and/or expansion in an organ-specific manner [32], including; JAGGED, AINTEGUMENTA, ARGOS, BPFp, OPR3, ARF8, BIG EROTHER, KLU and DAI.
Recently, various powerful techniques have been developed, including; microarrays, cDNA or Expressed Sequence Tag (EST) sequencing, Serial Analysis of Gene Expression (SAGE), which have provided the illustration of transcriptome dynamics in Arabidopsis, wheat and maize [33-35]. However, these data are far from being complete due to the limitations of these approaches. Hence next-generation high-throughput sequencing tools have emerged, offering high coverage at a low-cost. The RNA sequencing method (RNA-seq) represents the latest and most powerful tool for characterizing the transcriptome [36]. With sufficient sequencing depth and sensitivity, RNA-seq can produce ultra-high-throughput data and is more suitable for gene expression studies [36,37]. In addition to this powerful technology, the Chinese cabbage genome sequence has been released [38] and lays the foundation for our present sequencing studies.
In this work, we employed Chinese cabbage vegetative mass at the four-leaf stage and petals in their full-bloom stage as test materials. For analysis using next-generation high-throughput sequencing (RNA-seq), we isolated the RNA sequences from the two samples and successfully identified the associated differentially expressed genes. The RNA-seq analysis clearly provided extensive novel information about the Chinese cabbage transcriptome during petal development. Further functional analysis of the set of differentially transcribed genes will help us to elucidate the mechanism of petal development and may greatly facilitate the breeding practices at a molecular level.
RNA extracted of vegetative mass and petal
Inbred lines of Chinese cabbage used in this study were grown under greenhouse conditions. Two samples were selected at the four-leaf stage (i) seedling with roots (vegetative mass) and (ii) petals in full-bloom, respectively (Figure 1). For sequencing, total RNA was extracted from the two samples with Trizol, according to the manufacturer’s instructions. The quality and purity of each RNA were attained using Agilent 2100 Bioanalyzer and 1.0% agarose gel electrophoresis.

Figure 1: a. The four-leaf stage seedling with roots (vegetative mass); b. inflorescence in full-bloom; c. Petals in full-bloom.
Library construction and sequencing
After extraction of total RNA from the samples, mRNA was enriched by using magnetic beads coated with oligo (dT). Upon addition of the fragmentation buffer, the mRNA was converted to short fragments (~200 bp). Subsequently, the cDNA was synthesized by using the mRNA fragments as templates. The double strand cDNA was purified with the QiaQuick PCR extraction kit and washed with EB buffer for end repair and single adenine (A) nucleotide addition. Finally, sequencing adapters were ligated to the fragments. The required fragments were purified by agarose gel electrophoresis and enriched by PCR amplification. The library products were then transferred for sequencing analysis with Illumina HiSeq™ 2000. Importantly, base calling was used to convert the original image data into sequences, to acquire clean reads before further analysis.
Bioinformatics
DEG tag annotation: After sequencing, the base calling step was repeated to eliminate any unwanted raw reads with adaptors and low quality. This included unknown bases >10% and low quality reads (that were determined by a percentage value of ≤ 5, attributing to >50% in a single read). Subsequently, clean reads were mapped to reference sequences using the SOAPaligner/soap2 system [39], with the maximum two base mismatch allowance in the alignment.
Gene expression levels: The gene expression was calculated by the numbers of reads mapped to the reference sequence and each gene. The gene expression level was calculated by using RPKM method (reads per kb per million reads) [40]. The method was able to eliminate the influence of different gene length and sequencing discrepancy, based on the calculation of gene expression. Therefore, the calculated gene expression was directly used for comparing the difference of gene expression among our samples.
Screening of Differentially Expressed Genes (DEGs): The samples were analyzed to detect genes with varying expression levels. Referring to "the significance of digital gene expression profiles [41]”, we were able to successfully identify Differentially Expressed Genes (DEGs) between the two samples. This was conducted using the False Discovery Rate (FDR, ≤0.001) and the absolute value of log2 Ratio ≥ 1 (Ratio represented the fold of different expression), as the threshold to judge the significance of the gene expression difference.
Gene ontology analysis of DEGs: GO enrichment analysis provided GO terms that were significantly enriched in DEGs compared to the genome background, and filtered DEGs that correspond to biological functions. The method mapped all available DEGs to GO terms from the database (http://www.geneontology.org/), calculating gene numbers for each term. Using a hyper-geometric test, all highly enriched GO terms in DEGs were then compared against the genome background. The calculated p-value passing through Bonferroni Correction, were corrected to a p-value ≤ 0.05 and set as a threshold. GO terms fulfilling this condition were defined as significantly enriched GO terms in DEGs. This analysis was able to recognize the main biological functions that were exercised by DEGs.
Pathway enrichment analysis of DEGs: KEGG is the major public pathway-related database [42]. Pathway enrichment analysis was used to: (i) identify significantly enriched metabolic or signal transduction pathways in DEGs, and (ii) compare with the whole genome background. The “Q value ≤ 0.05” was used as the threshold to judge the significantly enriched pathway of differentially expressed genes.
Real-time quantitative PCR analysis: A selection of nine DEGs was taken from all present DEGs for analysis using Real-Time Quantitative PCR (qRT-PCR). For this step, the total RNA was treated with DNase I enzyme and subsequently converted to single strand cDNA, by the SuperScript® III First-Strand Synthesis System (Invitrogen), according to the manufacturers’ protocol. The specific gene primers were designed using Primer 5.0, according to the cDNA sequences in Table 1. Actin was used as the internal control to normalize small differences in template quantities. This was monitored using Bio-Rad iQ5 system and the data was collected using method of relative quantification 2-ΔΔCt.
| Bra014822-L | AGTTTATCAGCCCTAACACCACA |
| Bra014822-R | TCTGAGTCCGAAGATTTCTATTTGT |
| Bra020093-L | GATGACTGATTATTGTTGTCCTTCC |
| Bra020093-R | ATTGTATATCTTCCCCCTTCAAATG |
| Bra002285-L | TGACTGATTATTGTTGTCCTTCCAT |
| Bra002285-R | ACTGTATATCTTCCCCTTTCAAATG |
| Bra021470-L | CTTGGAGAGGACCTTGGACCC |
| Bra021470-R | CTTCATTGACAAAGCACGATTGG |
| Bra006322-L | AGGAGGATGGGAAGGAAATGAAC |
| Bra006322-R | TCTGGAATGTAACCGGGCTGTG |
| Bra004361-L | TTGGAGAGAAACCAGAGGCAC |
| Bra004361-R | GTTCCTGTATCGCCTTCTCCTT |
| Bra038326-L | CCATAAGGGGAAACTCTTTGAATA |
| Bra038326-R | CTGGTTTCTCTCCAAAAGGTCAA |
| Bra008674-L | CTGGATGATATGATTGGTGTGAGA |
| Bra008674-R | AAGAGGCTGGAATAGTCCTTGAG |
| Bra039170-L | TAGACGGCTCTCTGAAGCAAGTT |
| Bra039170-R | CCCTCCTACATGGTGACTCCTC |
| Actin-L | CCTCTTAACCCAAAGGCTAACAG |
| Actin-R | CATCACCAGAATCAAGCACAATAC |
Table 1: Primers.
Establishment of RNA library
Two independent RNA libraries for petal and vegetative mass were established and the Illumina HiSeq™ 2000 was used for further sequencing. The original image data was transferred into sequences by base calling. After removing the low quality reads and masking adaptor sequences, a total of 7,065,650 and 7,157,099 clean reads were obtained for the petal and vegetative mass samples, respectively. Clean reads were mapped to the reference genome using SOAPaligner/soap2. Mismatches up to 2 bp were permitted in the alignment. The petal sample gave a total of 5,725,584 mapped reads and the vegetative sample displayed 5,688,759 (Table 2).
| Stat. of Map to Genome | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Total reads | Total base pairs (bp) | Total mapped reads | Perfect match | ≤3bp mismatch | Unique match | Multi-position match | Total unmapped reads | ||
| Petal | Number of reads | 7065650 | 346216850 | 5725584 | 4406407 | 1319177 | 5415396 | 310188 | 1340066 |
| % | 100 | 100 | 81.03 | 62.36 | 18.67 | 76.64 | 4.39 | ||
| Vegetative mass | Reads number | 7157099 | 350697851 | 5688759 | 4115443 | 1573316 | 5269441 | 419318 | 1468340 |
| % | 100 | 79.48 | 57.50 | 21.98 | 73.63 | 5.86 | 20.52 | ||
Table 2: Statistical results of samples map to reference genome.
This was followed by quality assessment of the sequencing data. The raw results gave 99.47% clean reads in the petal sample, and the remainder was attributed to the either low quality or the adapter, containing N. For raw reads of the vegetative mass sample, clean reads accounted for 99.45% (Figure 2A and B). Finally, the statistical gene coverage (Figures 2C and D), were determined to reflect the quality of sequencing. From this, we found that the gene coverage from the two RNA libraries exhibited a high degree of repeatability and consistency indicating high quality sequencing results that was suitable for subsequent analysis.
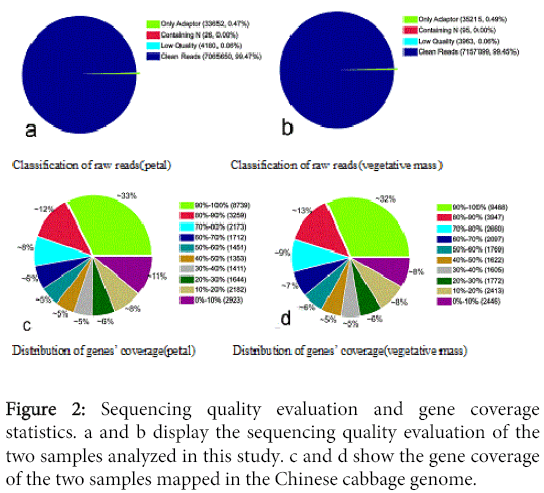
Figure 2: Sequencing quality evaluation and gene coverage statistics. a and b display the sequencing quality evaluation of the two samples analyzed in this study. c and d show the gene coverage of the two samples mapped in the Chinese cabbage genome.
Analysis of differential expression genes (DEGs)
For RNA-seq, analysis of DEGs between two samples was crucial. In order to analyze the different gene expression patterns, the Reads per Kilo Base per Million (RPKM) measure was used to calculate the abundance of expressed genes. We compared RNA libraries of our two samples, and observed 11,079 genes that displayed significant changes in expression. Within this, 3,900 DEGs were up-regulated in the petal sample, and 7179 DEGs were down-regulated.
We observed a large number of Specifically Expressed Genes (SEG) in these DEGs. SEGs defined as those that did not express in one library but the reads number >11 in another [43]. The results showed 1595 SEGs, in them 303 SEGs were up-regulated, including: PISTILLATA, APETALA3, SEPALLATA2, Sep1, homeotic protein and boil AP1. The remaining 1292 SEGs were down-regulated, such as the superoxide dismutase, PDF 1 (PROTODERMAL FACTOR1), pectate lyase, cytochrome P450 (Supplementary Table 1).
Gene ontology (GO) analysis of DEGs
In order to visualize the large amount of data generated by the RNA-seq, we used Gene ontology (GO) terms tool to obtain more information on the genes’ function. All DEGs used for GO analysis were divided into three sections (molecular function, cellular component and biological process). Biological processes contained the majority of GO annotations (1342; 63.5%), followed by molecular functions (568; 25.7%) and cellular components (202; 9.1%). Typically, the top five GO terms of the large cluster frequency in biological processes are: response to stimulus, macromolecule metabolic, protein metabolic, nucleic acid metabolic processes, and response to organic substance. Within function ontology, these terms comprise of: catalytic activity, nucleotide binding, hydrolase activity, metal ion binding, and purine nucleotide binding. Meanwhile cellular components are divided into cell, cell parts, intracellular, intracellular part and organelle.
Expression of flower development model genes
During the development of petals, the ABE type genes in the flower development model play an important role. In a larger set of DEGs, we found 10 genes that belong to this group, including; PISTILLATA, APETALA3, SEPALLATA2, SEP1. Importantly, the DEG analysis showed that majority of these genes were all expressed as up-regulated and exhibited high differential expression levels, with nine detected as SEGs (Table 4).
SEGs of unknown function
A large proportion of the 708 SEGs did not display explicit gene function. Of which 474 SEGs were hypothetical or putative proteins (Supplementary Table 3), with no confirmation of final function. Meanwhile, 353 of these genes were hypothetical protein ARALYDRAFT. Furthermore, apart from the hypothetical and putative proteins, the function of 86 (an additional SEG) were unknown (Supplementary Table 3). Therefore a more in-depth study is required to determine what role these genes play in petal development.
Real-time quantitative PCR validation of RNA-seq results
In order to confirm the results of RNA-seq, we selected nine genes for Real-Time Quantitative PCR (qRT-PCR). The results from these measurements are shown in Figure 3. Here we observe consistent data between the expression pattern obtained by qRT-PCR and RNA-seq, which proves the accuracy of the RNA-seq technique. Additionally, further qRT-PCR was conducted to identify the expression of these genes in pistil, anther, filament and pollen (Figure 3).
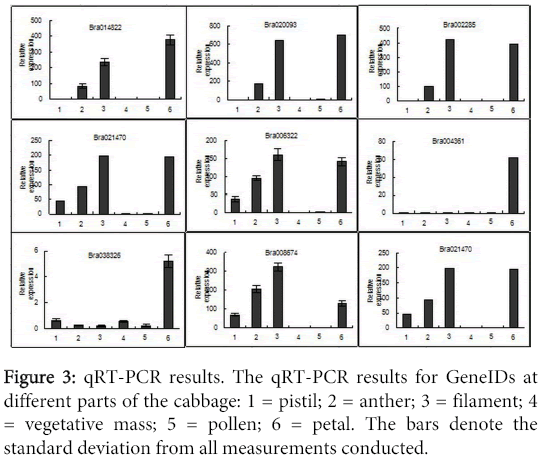
Figure 3: qRT-PCR results. The qRT-PCR results for GeneIDs at different parts of the cabbage: 1 = pistil; 2 = anther; 3 = filament; 4 = vegetative mass; 5 = pollen; 6 = petal. The bars denote the standard deviation from all measurements conducted.
In this study, we lay the foundation for petal development genes. By employing next-generation high-throughput sequencing technology, we identify differentially expressed genes between petal and vegetative mass in Chinese cabbage. The results are encouraging and reliable as demonstrated by qRT-PCR. We obtain a large number of differentially expressed genes, with specific selection expressed in petal. The present study not only supplements the knowledge of Brassica rapa ssp. pekinensis, but may also be used in research concerning the improvement of cabbage cultivation.
Flower development model genes
In previous studies of Arabidopsis floral organ, the organ types are typically defined as whorls 1-4. The development of every whorl can be controlled by different class genes, where the transcription factors are well identified; such as PISTILLATA (PI), APETALA1A (AP1), PETALA3 (AP3), SEPALLATA1-4 (SEP1-SEP4).
Nevertheless, the AP1 not only acts as the floral meristem identity genes, but can be used towards floral organ identity genes. In early floral differentiation experiments, it was expressed throughout the floral primordia. However in the formative stages of floral organs the AP1 was found to only express in sepals and petals. Consequently, two DEGs encoding the homeotic protein boi1AP1, were obtained with similar expression patterns; Bra004361 and Bra038326, with lengths of 570 and 771 bp respectively. Meanwhile, in the present study, the specifically expressed genes PI and AP3 (which are required in the development process) were both up-regulated in petals and were also found to express in stamen. Importantly, predecessors found in wild-type flowers that lacked the PI gene led to petal replacement by sepal and stamen replacement by carpel and filament. The study by Krizek et al. [19] reported that the plant flower organ with p35S-PI (a part of calyx) could transform into petals. In the case of AP3, this is commonly detected once the first whorl calyx primordial star is formed. Therefore, as the petal and stamen primordial emerge, AP3 RNA is present at considerably high levels across all cells of this organ [44]. Related experiments have shown that in flowering plants; p35S-PI and p35S-AP3, the first and fourth whorl transformations are close to completion than the corresponding partial transformations exhibited by p35S-PI (first whorl) or p35S-AP3 (fourth whorl) [19]. Furthermore, we find that the PI (n = 3) and 1 AP3 genes obtained in this study are members of the MADS-box family, are SEGs, and are up-regulated in petal. For the three PI genes detected, two display longer lengths (Bra006549, 627 bp; Bra020093, 612 bp), while a shorter; Bra002285 441 bp is attained. The most significant differences in gene expression are observed in the order; Bra006549 > Bra020093 > Bra002285.
As the in-depth study of flower development people found, if want to make the plant vegetative organs turn to flower organs also need to spend another floral-specific genes (class E). In Arabidopsis, the expression of the ABC class gene with SEP can transform blade into a complete floral organs, with the development of petals commonly determined by AP1, AP3, PI and a single SEP gene [10,23]. In our study, we found DEGs SEP-1 and SEP-2 with expression levels much higher in petals than in vegetative mass. In addition, no information was found to validate the SEP-3 in DEGs.
The SEP-1 and SEP-2 are members of the large family of type II MADS-box transcriptional regulators [45,46], and are considered to have functional redundancy, thus, knocking out one of them can affect the overall phenotype. Moreover, they both participate in the establishment of floral organ identity [18,22]. Therefore, if the flowering plant lacks SEP-1, SEP-2 and SEP-3 at the same time, all floral organs organizations are expected to convert to sepals [22]. Thus, they are the necessary genes to form the petals, stamen and carpel.
Other related genes
From the differentially expressed genes study we also detected the MYB and several other MYB-related genes. Of which 17 were up-regulated and 39 were expressed as down-regulated. Previous studies of have demonstrated that the MYB gene function plays a key role in the process of plant growth and development [47]. The MYB gene family is vast, and subsequently involved in many metabolic processes including; plant secondary metabolic processes, regulating cell morphology (involved in the response to plant biotic and abiotic factors). For example, a branch of this gene as a transcription factor involved in phenylpropanoid metabolism, and produces the main plant pigmen-anthocyanin. Additionally, the study relating to corn, the R2R3MYB transcription factor (a regulatory protein) has widely been involved in the regulation of the phenylpropanoid metabolic pathway, resulting in color variation within its seed, coleoptile, root, stem, leaf and male flower spikes [48-51]. MYB can also influence the cell morphology. For example, the MIXTA gene of snapdragon and its homologous morning glory gene PhMYB1, are indispensable in the regulation of conical shape cell development in the petals’ epidermis [52]. Moreover, Byongchu’s study has reported on the ectopic expression of AtMyb21 which induces a series of morphological variation, including the reduction in size for leaves and petals of the transgenic plant [53]. This suggests that the MYB is an important gene which can affect the petal color and form.
In addition to the above genes we also obtained DEGs that may be related to the formation and development of petals in this study, including the APUM 12, plastid-lipid associated protein PAP 2, RTFL 5. It is assumed that a combined action of all these genes results in the generation of petals and more worthy of our attention are the SEGs of unknown function. Therefore, our future studies aim to identify and investigate the functions of these crucial genes and discover their association with petal development and provide insights into their mechanisms.
Liu Chang and Liu Zhiyong contributed equally to this work.