Journal of Physical Chemistry & Biophysics
Open Access
ISSN: 2161-0398
ISSN: 2161-0398
Research Article - (2016) Volume 6, Issue 2
5-Fluorouracil is clinically utilized as antitumor drug to treat numerous sorts of malignancy, which is made accessible to the objective tissues in conjugation with transport protein serum albumin furthermore which is low harmful when compared to the other drugs of this family and hence its binding characteristics are therefore of prime interest. The steady state and time resolved fluorescence studies, Fourier transform infrared spectroscopy and circular dichroism studies were employed to explain the mode and the mechanism of interaction of 5FU with HSA. 5-Fluorouracil binding is characterized with one high affinity binding site, with the binding constant of the order of 104. The molecular distance r (1.23 nm) between donor (HSA) and acceptor (5-FU) was estimated according to Forster's theory of non-radiative energy transfer. The feature of 5-Fluorouracil induced structural changes of human serum albumin has been studied in detail by Raman spectroscopy, circular dichroism and Fourier transform infrared spectroscopy analysis. The binding dynamics was expounded by synchronous fluorescence spectroscopy, fluorescence lifetime measurements and molecular modelling elicits that hydrophobic interactions and hydrogen bonding, stabilizes the 5-Fluorouracil interaction with HSA.
<Keywords: Human serum albumin; Fluorescence quenching; Anisotropy; Energy transfer; Molecular modeling
Malignancy is standout amongst the most driving reason of adult deaths worldwide. Around 15% of all incidents cancers worldwide are attributable to infections. This percentage is about three times higher in developing countries (26%) than in developed countries (8%) (American cancer society) [1]. For several decades, chemotherapy regimens based on the drug 5-Fluorouracil (Figure 1) is widely recognized as effective treatment modality, especially with tumors. It has been used to treat several types of cancers including colon, rectum, and head and neck cancers, also which exhibits antibacterial activity [2].

Figure 1: Chemical structure of 5-Fluorouracil.
Research on molecular interactions of drugs with biological molecules has greatly contributed to explain the structures and functions of bio macromolecules such as proteins, deoxyreboneuclotide and some biophysical processes. By binding to human serum albumin (HSA), most drugs circulate in plasma and reach the target tissues and their distribution is mainly controlled by HSA. Therefore, drug binding to proteins has become an important determinant of pharmacokinetics.
Serum albumins are the major soluble protein constituents of the circulatory system and have many physiological functions, including acting as transporters for numerous endogenous and exogenous ligands (e.g., drugs, fatty acids, etc.). Drug binding to serum albumin is an important determinant of drug pharmacokinetics, restricting the unbound concentration and affecting drug distribution and elimination [3]. Human serum albumin is the major protein component of blood plasma (approx. 60% of the total protein) [4] It is a single, non-glycosylated polypeptide that organizes to form a heart-shaped protein with approximately 67% α-helix [5]. HSA is capable of binding reversibly a wide variety of drugs, resulting in an increased solubility in plasma, decreased toxicity, and/or protection against oxidation of the bound ligand. The protein’s capability of binding aromatic and heterocyclic compounds largely depends on the existence of two major binding regions, namely Sudlow’s sites I and II [6-9] Site I, also known as the warfarin binding site, is formed by a pocket in subdomain IIA and contains the only tryptophan of HSA (Trp 214). Site II is located in subdomain IIIA and is known as the benzodiazepine binding site [10]. Of these sites, site I seems to be the more versatile, because it can bind, with a high affinity, to ligands that are very different from a chemical point of view [11]. Recently, subdomain IB of HSA consist of hydrophobic groove formed by alpha helices H7, H8, H9 covered by loop L1. This hydrophobic groove created two access site to bind drug molecules (Camptothecin and 9-amino camptothecin) termed as distal site (IBd between loop L1 and alpha helices H7 and H8) and proximal site (IBp between L1 and alpha helix H9) [12].
The different aspect of photo physical interaction can be used to probe the nature of the binding due to drugs with bio macromolecules. In this context, we discussed the 5-Flurouracil with human serum albumin protein conformational changes using various spectroscopy techniques, UV-Visible, Fluorescence emission spectroscopy, synchronous fluorescence spectroscopy, Fluorescence resonance energy transfer (FRET), Raman spectroscopy and circular dichroism (CD) spectroscopy. To reach this goal we have used both biophysical and computational approaches and emphasized the mode of binding 5-FU with HSA.
Materials
Human serum albumin was purchased from Sigma Aldrich, USA and it was used without any further purification. Millipore water was used for preparing solution throughout the experiments. HSA solution was prepared in Tris-HCl buffer solution of pH 7.4. HSA solution was kept in the dark at 4°C. 5-Fluorouracil (≥ 99%) was purchased from Celon Laboratories Ltd., Hyderabad, India, and the stock solution of 5-FU was also prepared using the same buffer.
Methods
Absorption spectra were recorded in the wavelength range of 225-400 nm using UV-Visible absorption spectrophotometer (Perkin- Elmer Lamda35, Waltham, MA). The steady state fluorescence emission measurements were made using a commercially available spectrofluorometer (Fluoromax-2, ISA; Jobin-Yuvon-Spex, Edison, NJ) and spectral band pass was kept as 5 nm for both excitation and emission monochromators. The emission spectrum was recorded in the wavelength region 300-440 nm at 280 nm excitation. Synchronous fluorescence spectra were recorded by simultaneously scanning the excitation (λex) and emission (λem) monochromators with two different constant wavelength intervals (Δλ) such as 15 nm and 60 nm between the excitation and emission monochromators. The Excitation emission matrices were mapped with the following conditions: the emission wavelength was recorded between 200 and 750 nm, the initial excitation wavelength was set to 200 nm with an increment of 20 nm. The numbers of scanning curves were 26 and other scanning parameters were identical to fluorescence emission spectral measurements. In EEM measurements, the spectral band passes were kept as 5 nm for both excitation and emission. The anisotropy values were measured by using Fluoromax-2 equipped with a excitation source (150 W ozone free xenon arc lamp) coupled to the monochromator delivers light to the sample at a desired wavelength and the fluorescence emission from the sample is collected by an emission monochromator to a photomultiplier tube (R928; Hamamatzu, Shizuoka-Ken, Japan). In addition, using the absorption spectrum of 5-FU and fluorescence spectrum of HSA, (Concentration ratio of drug and protein is 1:1 at pH 7.4) the fluorescence resonance energy transfer and energy transfer efficacy of HSA with various concentration of drug also evaluated.
Lifetime measurements were made using Time Correlated Single Photon Counting System (TCSPC, Horiba Jobin Yuvon IBH, UK) with a fast response red sensitive PMT (Hamamatsu Photonics, Japan) detector. The excitation source used in the experiments was a light emitting diode laser of wavelength 280 nm (pulse width <0.1 ns) and the decay measurements were made at 350 nm. The fluorescence emission was collected at 90 degree from the path of the light source. The electrical signal was amplified by a TB-02 pulse amplifier (Horiba) fed to the constant fraction discriminator (CFD, Phillips, The Netherlands). The first detected photon was used as a start signal by a time-to-amplitude converter (TAC), and the excitation pulse triggered the stop signal. The multichannel analyzer (MCA) recorded repetitive start-stop signals from the TAC and generated a histogram of photons as a function of time-calibrated channels. The instrument response function was obtained using a Rayleigh scatter of Ludox-40 (40 wt. % suspension in water; Sigma-Aldrich) in a quartz cuvette at 280 nm. Decay analysis software (DAS6 V6.0, Horiba) was used to extract the lifetime components. The goodness of fit was judged by chisquare values as well as visual observations of fitted line and residuals. Each fluorescence decay was analyzed with two and three lifetimes and then values of χ2 were compared to determine the best fit. The time calibration of the system was 5.56 × 10-11 sec/ch.
The Raman spectrum was carried out using Lab Ram HR 800 using a Helium laser of wavelength 633 nm. The laser power at the sample measuring was 12 ± 0.5 mW focused to spot size of 1 μm. the instrument is equipped with an 800 nm Czemy-Tumer spectrograph and 1024 × 256 pixel Peltier cooled CCD. The calibration of the system was carried out for 520.7 cm-1 spectral line of silicon. The acquisition time was set to 30 sec and remove cosmic rays there spectra were acquired twice at every point. Every spectrum was corrected for non-uniform spectral response of the detection system, subtracted for Savitsky-Goly filter was applied to smooth the spectra.
Circular Dichroism (CD) measurements of HSA in the presence and absence of 5FU were made in the UV-region (200-260 nm) on a spectropolorimeter (Jasco J715, MD, USA) using a quartz cell of 0.1 cm. The CD profiles were obtained employing a scan speed 20 nm/min. Appropriate baseline corrections in the CD spectra were made. All the spectra were recorded at room temperature.
The X-ray crystal structure of Human Serum albumin (PDB ID: 4L8U) and 5-FU (PUBCHEM ID: 24278439) was taken for molecular docking studies. Structures were energetically minimized using OPLS2005 force field in impact module and subsequent minimized structure was used for flexible docking approach using Induced fit module (Glide) of Schrodinger 2014 [13]. Binding mode of 5-FU with HSA was analyzed in terms of binding energy (Glide Energy).
Docked complex of 5-FU with HSA and as well as free drug free HSA was used for molecular simulation study for comparison of stability and calculation of binding free energy using Amber12. To prepare the initial system for simulation, the free HSA and their 5-FU complex was protonated and solvated using TIP3P water box with a dimension of 10 Å and counter ions were added appropriately to make the total charge zero. The initial coordinates and corresponding topology files was created using molecular mechanics (99SB) force field for protein and using antechamber for 5-FU drug. The free HSA and their 5-FU complex system were relaxed with the help of two phase of energy minimization. The first phase of energy minimization was carried with 1000 cycle of steepest descent and 1500 cycle of conjugate gradient with solute atom restrained by a harmonic potential with force constant by 10 Kcal/molÅ2. The second phase of energy minimization was carried out using conjugate gradient method for 2500 cycle for full system minimization without any restraint.
Further, the two phase of equilibration, first with positional restraint on solute atom (protein and 5-FU) and other one without restraint were run for bringing the temperature and pressure of each system with 300 K and 1 atm respectively for making isothermal-isobaric ensemble system. The temperature and pressure of each system was stabilized around 300 K using Barendsen temperature coupling (Thermostat) and langevin barostat respectively. The equilibration was done for 1 ns with 2 fs integration time step, all atoms belonging solute atoms was restrained by harmonic potential with force constant 10 kcal/molÅ2. SHAKE algorithm was used to constrained hydrogen linked bonds. The equilibrated system with periodic boundary condition was used to perform constant temperature and pressure simulation for 15 ns with 1 fs integration time step using leag-frog integrator. Simulation trajactory was saved on 1 ps of simulation time and further used for structural analysis. The MD simulation was performed on 8-GPU cluster built in CAS in Crystallography and Biophysics University of Madras, Chennai.
The binding free energy of 5-FU with HSA was done with help of MM-GBSA method which are implement in MMPBSA.py script of AMBER12. The first step in MM-GBSA is to generate multiple snapshots from the stable MD production trajactory of the 5-FU HSA complex, Here 300 snapshots were collected, equally spaced at 100 ps intervals. For each snapshot, a free energy is calculated for each molecular species and the ligand binding free energy is estimated as follows
UV-Visible absorption spectroscopy studies
The UV-Vis absorption characterization is an applicable method to know the complex formation between small molecules and proteins. Human Serum Albumin has the absorption sort band at 280 nm, Figure 2(a) shows the UV-Vis absorption spectra of HSA-5FU system in the presence of Fluorouracil. The absorbance of HSA is formed to increase with increase of the concentration of 5-FU. Further it is observed that there is a blue shift in the maximum peak position for HSA - 5-FU systems, indicating that there is a strong interaction of 5-FUand HSA complex may form, the observed changes in the absorbance of HSA-5- FU complexes have indicated that an interaction occurred between these drug and HAS (Figure 2b); and the considerable blue shift in maximum peak positions may be accounted for the change in polarity around the tryptophan residue and hence changes in hydrophobicity in both HSAdrug complexes [13-15]. Beside of UV-Vis absorption spectroscopy, fluorescence spectroscopy is another powerful method to study the interactions between small molecules and bio macromolecules.

Figure 2: The UV absorption spectra of HSA varying with the concentration 5-FU in the Tris-HCl buffer solution (pH 7.4) at room temperature (25°C). (a) Concentration of HSA (1 × 10-5 M) (1). Concentration of 5FU 0-45 μM with 5 μM interval (2-10). (b) Difference spectrum.
Steady state/time resolved fluorescence spectroscopy studies
Fluorescence quenching was used to evaluate the structural changes of HSA when interacting with 5-FU. The intrinsic fluorescence nature is mainly due to the tryptophan more than 95%, which is located in the 214 sequence position [9]. The fluorescence spectra of HSA with different concentration of 5-FU were measured shown in Figure 3(a). The fluorescence intensity of HSA decreased remarkably with increasing concentrations of 5-FU and a blue shift was observed, which indicated that 5-FU interacted with HSA and the fluorescence chromophore of HSA was placed in a more hydrophobic environment after adding 5-FU. To demonstrate the quenching mechanism, the fluorescence quenching data were analyzed using the well-known Stern-Volmer equation [16]

Figure 3: (a).Fluorescence emission spectra of HSA-5-FU system (pH = 7.4). Concentration of HSA (10 μM). Concentration of 5FU 0-65μM with 5μM interval (2-14). (b).Stern Volmer plot. (c) The time resolved fluorescence decay profiles of HSA in absence and presence of 5-FU in Tris–HCl buffer of pH 7.4 various concentration of λex=280 nm and λem=345 nm. HSA concentration was fixed at 10μM. In (1–3) 5-FU concentrations were of 0, 10 and 80 μM. (d) The plot of log [(F0–F)/F] versus log [Q].
F0/F=1+Kq τ0[Q]=1+Ksv[Q] [1]
where F0 and F are the fluorescence intensity in the absence and presence of quencher, respectively; Kq stands for the quenching rate constant of the bio macromolecules τ0 is the average lifetime of the bio macromolecules without quenchers with a value of 2 × 108. KSV is the Stern-Volmer quenching constant and [Q] is the concentration of quencher. The Stern-Volmer plots for the quenching of HSA by 5-FU is shown in Figure 3(b). Fluorescence quenching of HSA was initiated by complex formation between HSA and 5-FU rather than by dynamic collision between the two substances. The static quenching mechanism can also be proved by the fact that values of Kq were greater than the limiting diffusion rate constant of the biopolymer (1.47 × 1013 L-1 mol-1 s-1).
To confirm the exact quenching mechanism, time-resolved fluorescence analysis was performed. Figure 4 shows the fluorescence decay of HSA in the absence and presence of 5-FU. HSA exhibits single exponential decay not only in dilute solutions but also in the presence of 5FU. While increasing the concentration of 5-FU there is considerable changes in the lifetime of HSA (from 4.23 ns to 2.47 ns). This observation shows quenching follows a static mechanism and also supports the formation of a ground state surface complex (Table 1).

Figure 4: Synchronous fluorescence spectra (a).Δλ=15, (b). Δλ=60 nm HSA (10μM)in the presence of 0,10,15,20,25,30 μM 5-FU respectively.
| Fluorescence Decay Fitting Parameters for HSA-5Fluorouracil (pH 7.4) | ||||||||
|---|---|---|---|---|---|---|---|---|
| [5-FU] (10-6M) | α1 ±(Error×10-3) | α2 ±(Error×10-3) | α3 ±(Error×10-3) | τ1 (ns) | τ2 (ns) | τ3 (ns) | 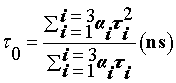 |
Χ2 |
| 0 | 0.44 ±0.36 | 0.08 ±0.76 | 0.47 ±0.10 | 4.30 ±0.28 | 1.00 ±0.12 | 11.7 ±0.16 | 4.52 ±0.19 | 1.07 |
| 10 | 0.59 ±0.26 | 0.13 ±1.33 | 0.46 ±0.14 | 4.79 ±0.27 | 1.09 ±0.04 | 6.03 ±0.01 | 2.73 ±0.11 | 1.03 |
| 80 | 0.37 ±0.36 | 0.28 ±1.67 | 0.35 ±0.10 | 2.67 ±0.22 | 0.34 ±0.03 | 7.85 ±0.18 | 2.23 ±0.14 | 1.08 |
Table 1:Florescence lifetime profile of HSA and increasing concentration of 5-FU.
Binding constant and the number of binding sites
The binding constant ka and the number of binding sites can be calculated using the following equation [17].
Log [(F0/F)/F]=LogKa+nlog [Q] [2]
A plot of log [(F0-F)/F] Vs log [Q] gives a straight line (Figure 5). From the plot we can obtain the binding constant K=2.89 × 104 Lmol-1 and binding site n=0.98. The results indicate that there is one class of binding site for drug in HSA and drugs could be bound and transported by HSA to the body.

Figure 5: Three-dimensional fluorescence contour map of HSA (a) and 5FUHSA system (b).
Synchronous fluorescence spectroscopy
Synchronous fluorescence spectra can provide valuable information about the molecular microenvironment in the vicinity of the fluorophore functional groups. The Δλ values (scanning interval, Δλ=λem-λex) stabilized at 15 or 60 nm, synchronous fluorescence of HSA gives the characteristic information of tyrosine and tryptophan residues, respectively [18]. In this study the synchronous fluorescence spectroscopy was used to get the information about the binding sites of 5-FU to human serum albumin molecule. In the synchronous fluorescence spectra of HSA, the shift in position of maximum emission wavelength corresponds to the changes of polarity around the fluorophore of amino acid residues.
As can be seen from Figure 6, the fluorescence of tyrosine residues is weak and the position of maximum emission wave-length showed little change when Δλ was 15 nm. While the fluorescence of tryptophan residues was strong and the position of maximum emission wavelength shifted moderately toward lower wavelength when Δλ was 60 nm. This reflects the fact that the microenvironment of the tryptophan residues was significantly affected by 5-FU binding. Furthermore, the blue shift effect expressed that the conformation of HSA was changed. It is also indicated that the polarity around the tryptophan residues was decreased while the hydrophobicity was increased.

Figure 6: Steady state anisotropy values of 5-FU at 444 nm and increasing concentration of HSA. Concentration of 5-FU 20 × 106 M and concentration of HSA in the mixture is 1 to 60 × 106 M.
Three-dimensional fluorescence spectroscopy
The three dimensional spectra is a powerful method to study the conformational changes of protein and also it can provide total information regarding the fluorescence characteristics by changing the excitation emission wavelength simultaneously. The three dimensional florescent spectra and the contour maps of HSA and HSA- 5FU complex are shown in Figure 7a and 7b respectively. As shown in the figure, the fluorescent intensity of peak decreased with addition of drug (5FU) in a system and the possible reason is due to complex formation between drug and protein. The result indicates that fluorescent intensity of HSA-5FU complex was lower than that in free HSA, in which the polarity around both residues, Trp and Tyr may be reduced. This means that the binding site between HSA and the drug is located within this hydrophobic pocket. The decrease of fluorescence emission intensity of this peak together with the synchronous fluorescence result reveals that the interaction of drug (5FU) with HSA induced some changes in the microenvironment and the confirmation of protein [19].

Figure 7: The overlap of the fluorescence emission spectrum of HSA (a) and the absorption coefficient spectrum of 5-FU.([HSA]:[5-FU]=1:1), pH=7.4.
Steady state fluorescence anisotropy
Fluorescence anisotropy provides useful information on the change in orientation of a small molecule upon binding with macromolecules such as protein and DNA. The interaction of 5FU with HSA was also characterized by studying the fluorescence anisotropy. Fluorescence anisotropy was measured for 5-FU with HSA of concentration ranging from 1-60 μM and the fluorescence anisotropy, r, was measured as follows: [19]
r=(IVV-GIVH)/(IVV+2GIVH) [3]
Where, IVV and IVH were the intensities obtained by fixing the excitation polarizer orientation vertically and changing the emission polarizer orientation vertically and horizontally, respectively.
The G factor is defined as
G=IHV/IHH [4]
Where, IHV and IHH were the intensities obtained by fixing the excitation polarizer orientation horizontally and changing the emission polarizer orientation vertically and horizontally, respectively. G is the factor that corrects the unequal transmission by the diffraction gratings of vertically and horizontally polarized light.
The anisotropy values of fluorophores in buffer or in any aqueous medium are very low, the fluorophores can freely rotate, whereas r values increases in motion restricted environments. For example it is reported that, anisotropy value for small molecules may be zero, for randomly oriented molecule and it increases to a maximum value of 0.4 when there is no rotation of molecule. Figure 8 shows the variation of r values of 5FU versus increasing concentration of HSA, which shows a sharp rise in the anisotropy value of 5FU up to 10 μM concentration and there after reaches a steady value and attains a maximum (r=0.13) at 40 μM HSA environment. This high anisotropy value may be due to the locking of 5-FU in motion restricted sites of HSA and may also be due to hydrogen bonding interaction between the amino acid residues of polypeptide backbone of HSA and -NH,-OH groups of 5-FU.

Figure 8: Raman spectra of HSA and HSA-5FU Complex.
Measurement of fluorescence energy transfer
5-FU can quench the intrinsic fluorescence of HSA, which suggests the occurrence of energy transfer between HSA and 5-FU. The fluorescence resonance energy transfer (FRET) includes radiation energy transfer and non-radiation energy transfer. Radiation energy transfer occurs when the fluorescence spectra of the donor is malformed. According to the Forster theory of non-radiation energy transfer energy transfer is likely to happen if the following conditions are met: (1) the donor can produce fluorescence light, (2) fluorescence emission spectra of the donor and UV-vis absorption spectra of the acceptor have enough overlap, (3) the distance between the donor and the acceptor is less than 7 nm.
The energy transfer efficiency E, the distance r and the critical energy transfer distance R0 are given by [20,21].
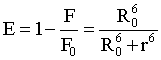 (5)
(5)
Where r is the distance between the donor and acceptor, R0 is the critical distance between the donor and acceptor when their transfer efficiency is 50%:
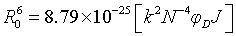 (6)
(6)
Where K2 is the spatial orientation factor of the dipole, n is the refractive index of the medium, F is the fluorescence quantum yield of the donor, and J is the overlap integral of the fluorescence emission spectrum of the donor with the absorption spectrum of the acceptor, which can be calculated by
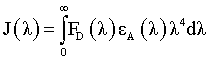 (7)
(7)
Where FD (λ) is the fluorescence intensity of the donor at wavelength λ to λ +Δλ, with the total intensity normalized to unity and εA (λ), the molar extinction coefficient of the acceptor at wavelength (λ). Figure 9 shows considerable overlap to the fluorescence emission spectrum of HSA with absorption spectrum of 5FU. From the overlapping of the absorption spectrum of the acceptor and fluorescence spectrum of the donor we can calculate the J value. That is J=3.5038 × 10-21 M-1 cm3. Under the experimental conditions we found R0=2.0087 nm from equation (2) using following values E=0.9485, k2=2/3 N=1.336 and ΦD=0.118, r=1.23 nm. Both R0 and r value is lower than the maximum academic values for R0 (5-10 nm) and the donor and acceptor distance r<7 nm.

Figure 9: CD spectra of HSA in the absence (1) and presence (2–3) of 5-FU (Con. of HAS 10 μM, Con. of 5-FU 10, 20 μM).
Micro Raman characterization
Raman spectroscopy can give a useful contribution since it can define small regions in large macromolecular complexes. For example, it can give a global insight into the overall secondary structural changes of proteins and information on some functional moiety, such as -SH, S-S, etc. and amino acid residues as Tyr, Trp, Phe and His [22]. One of the greatest advantages of Raman spectroscopy is the lack of sample preparation needed prior to analysis. Because Raman scatter originates from the surface of a sample (typically no deeper than several hundred microns), there is no concern with sample thickness, size or shape. Further advantages are a short measuring time, the low amount of sample required and the sample form can be solid, liquid, gel, etc.
HSA contains 31 Phe, 18 Tyr and 1 Trp residues that give rise to many Raman bands due to aromatic ring vibrations. Figure 10 shows Raman spectra of free HSA (black colour) and HSA - 5FU complex (red colour). As regards Trp, the intensity decrease in the shoulder in the 1200-1400 cm-1 range (Figure 10) could indicate a change in the Trp environment. The spectral region of Amide I band nearly 1600 cm- 1(C=O stretch). From the Raman spectrum we absorbed that there is some biochemical environmental changes in above mention spectral regions due HSA interacting with 5FU, which alter the structure of HSA.

Figure 10: Hydrogen bond interactions of ligand with the residues represented in green colored lines and nearby hydrophobic residues in red colored lines. In ligplot, ligand is represented in sticks.
Circular dichroism spectroscopy
Circular dichroism (CD) spectroscopy is a well-developed method in structural biology, with a growingly wide range of applications. It is a technique that plays an important role in the characterization of proteins, particularly for secondary structure (e.g., α-helix, β-sheet) determination. The CD spectrum of HSA has two negative bands at 208 nm and 222 nm, which represents the transition of π-π* and n-π* of α-helix structure.
To quantify the results, the mean residue ellipticity (MRE) in deg cm2dmol-1 and the α-helix contents of free and bound HSA were calculated using the following respective equations.
MRE = θ/Cpnl × 10 [8]
Where θ is the observed (Experimental) dichoism in milli degree, Cp is the molar concentration of the HSA, n is the number of amino acid residues in protein (582) and l is the optical length of the cell (0.1 cm).
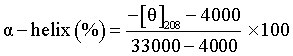
Where MRE208 is the detected Mean residual value at 208 nm, the value of 4000 is MRE value of the β-form and random coil conformation cross at 208 nm and 33000 is the MRE value of a pure α -helix at 208 nm.
The Figure 11 is the CD spectra of HSA and HSA with 5-FU. From the figure it is observed that there are two negative bands at 208 and 222 nm. These bands are contributed to n - π* transfer for the peptide bond of α-helical structure of protein. Using the equation 9 and 10, the α-helical content of free and bound HSA at two different concentration of 5FU were calculated. From the calculation it is bound that the percentage of α-helicity is decreased with increase of 5FU concentration. For example the percentage of helicity for pure HSA is found to be 53.31% and that in the presence of 5FU is found to be 48.40%, indicating that the binding of 5FU with HSA causes some conformational changes. It was reported that the change in the α-helicity due to the binding of drug at the hydrophobic surface of HSA. It was also reported that such type of interaction may not alter the secondary structure of protein (α-helix) which is important requirements for biomedical applications [23-26].

Figure 11: Molecular dynamic simulations of HSA complex with 5FU (Black: Free HSA Red: 5FU-HSA complex).
Molecular docking and simulation
Molecular modelling is very fruitful for understanding the binding interaction of 5-FU with HSA at atomistic level. Results from above fluorescence quenching and spectroscopic techniques suggest that 5-FU have strong binding affinity against HSA with a binding constant of K=2.89 × 104 Lmol-1. To understand the mechanism of the binding computational analysis were carried out. Thus we carried out computational studies to analyse the binding mode of 5-FU with HSA using molecular docking (Glide) and dynamic simulation, which helps us in understanding stability of protein-ligand complex in terms of thermodynamics parameter and docking result of 5-FU with HSA suggest that drug 5-FU favourably binds to lower region of proximal site in sub domain IB and interact with alpha helices H7, H8 and H9 with binding energy -25.45 Kcal/mol (in term of Glide energy) [27-29]. Uracil ring of 5-FU showed a π -π interaction with Phe149 and His146, hydrogen bond with Tyr 138 and many hydrophobic interactions with HSA (Figure 12; Table 2).

Figure 12: Binding free energy of Active site Residues.
| Compounds | Hydrogen bond(D-H...A) | Distance (Å) | Docking score (Kcal/mol) | Glide energy (Kcal/mol) |
|---|---|---|---|---|
| 5-FU | (Tyr 138)O-H...O | 2.73 | -4.43 | -25.79 |
| (HIS 146)N-H...N | 3.08 |
Table 2: Glide energy, docking score and hydrogen bond interaction of the ligands with key residues.
Simulation results of docked complex of drug 5-FU with HSA and as well as free HSA were analysed in terms of potential energy, structural deviation of overall fold of HSA and residual fluctuation. Root mean square deviation (RMSD) and radius of gyration (RGYR) were calculated for each conformation of 5-FU with HSA and compared with free HSA with respect to simulation (Figure 13a and 13b) time suggests that structural restriction occurred in HSA due to binding of 5- FU in subdomain IB and doesn’t distrupt the overall folding of HSA as represented by radius of gyration and suggest that complex are stable during simulation time. Residual fluctuations in backbone atoms (N, Cα and C) of 5-FU complex with HSA suggest smaller conformation fluctuation in active site residues occurred compared to that of drug free HSA (Figure 13c).
Binding of 5-FU with HSA was further analysed in terms of hydrogen and hydrophobic interaction occupancy during simulation time. 5-FU forms strong hydrogen bonding with Ser193 with 81% occupancy in last 1 ns of 15 ns simulation time (1000 conformation). And it also form π-π interaction with Phe149 with occupancy (66.63%) and hydrophobic interaction with the following residues, Gly189 (93.81%), Lys190 (60.54%), Ile142 (21.88%) and Arg184 (0.4%). Similarly the binding free energy of the active site residues had been plotted (Figure 14) suggesting His 146, Phe 149, Lys190 and Ser193 are more stable binding residue with ligand 5-FU.
Average binding free energy of HSA- 5-FU complex is -10.85(+/- 2.79) Kcal/Mol (without entropy contribution) calculated from the ensemble of simulated conformation using MMGBSA computational approach. Binding of 5-FU with HSA have almost equal electrostatic and vanderwaal energy contribution -17.40(± 1.68) to binding free energy.
The interaction between 5-FU and the protein HSA has been studied by computational studies and multi-spectroscopic methods. The fluorescence quenching phenomena of HSA depends on the concentration of 5-FU. It exhibits static quenching mechanism at experimental concentration range. The above results were also confirmed by steady state and time resolved fluorescence spectroscopy methods. From synchronous spectra it is established that the microenvironment close to both the tyrosine and tryptophan residues of HSA are disturbed. Steady rise in anisotropy of drug in the presence of HSA concludes the tight binding of the drug in the restricted hydrophobic pocket of the protein secondary structure. From the Forster non-radiative energy transfer study it has been found that the distance of 5-FU from HAS was 1.23 nm which predicts the possibility of energy transfer from the Trp donor to 5-FU acceptor. The results of micro-Raman, CD studies indicate that the secondary structure of the protein is not perturbed upon 5-FU binding over the concentration range studied. The experimental and modelling results were in good agreement and suggest that 5-FU could bind HSA through the hydrophobic force and hydrogen bonding. This binding study of 5-FU with HSA is of paramount importance in understanding chemicobiological interactions for drug design, pharmacy and biochemistry without altering the original structure. From the computational studies, understanding the albumin binding interaction with 5-FU in atomic detail and can be used this information together with knowledge of the drug target to design improvements, such as, lowering the effective dose and/or improving the efficacy of the target drug. To advance the understanding of the specific nature of human serum albumin drug transport for the purposes of drug design and development.
This work was supported by a Japan Society for the Promotion of Science (JSPS) Postdoctoral Fellowship from the Ministry of Education, Culture, Sports, Science and Technology.