Andrology-Open Access
Open Access
ISSN: 2167-0250
ISSN: 2167-0250
Research Article - (2024)Volume 13, Issue 5
High-performance liquid chromatography-tandem mass spectrometry has been pivotal to recent clinical advancements in the detection and monitoring of the progression of different metabolic disorders. Such systems have been used to routinely monitor serum testosterone levels in patients, to both mitigate side effects of different medications, as well as diagnose androgen-associated disorders. The present study develops and validates an LC-MS/MS-based assay for the detection of total serum testosterone from only (100 μL) of sample volume. To increase the high throughput capability of the developed diagnostic kit for clinical use on patient samples, the proposed method was streamlined via semi-automation on a JANUS® G3 Workstation liquid handler. The proposed diagnostic kit showcased high detection precision (CV<5%) as well as sample accuracy (93%-108%), within a clinically relevant range of testosterone serum concentrations (5-1500 ng/dl). Moreover, the assay showcased LOD of 5 ng/dl, which is significantly comparable to commercially established diagnostic tools. The developed method also exhibited high linearity over the specified testosterone serum range, with an obtained linearity of R2>0.99 using both manual as well as automated sample preparation. Overall, the work shown presents an optimized, automatable LC-MS/MSbased testosterone diagnostic assay for the routine evaluation of patient testosterone levels in serum for clinical and research use.
Testosterone; Mass spectrometry; Liquid chromatography; Sample preparation; Targeted metabolomics
The measurement of total Testosterone (T) in human serum at very low levels is pivotal in the clinical practice of diagnostics, treatment, and prevention of a variety of sex-hormone related disorders. This includes the diagnosis of hypogonadism and androgen deficiencies in men and Polycystic Ovarian Syndrome (PCOS) in women, as well as various types of cancers. Abnormally excessive or deficient T levels in patients may be associated with doses and side effects of patient medications and prescriptions [1-6]. Hence, clinical diagnostic and therapeutic studies require accurate monitoring of patient T levels, to ensure both patients’ improved long-term healthcare and quality of life.
Advances have been made in the field of quantification and screening of low or elevated T levels using immunoassays [7]. However, such assays suffer from notable discrepancy in results that limit their utilization within wider diagnostic uses [8]. Due to its increased detection specificity when detecting analytes at significantly lower concentrations Liquid Chromatography (LC) Tandem Mass Spectrometry (MS) approaches were alternatively investigated in the screening of patient T levels [9-11]. Despite being widely studied, limitations exist specifically in terms of repeatability in regard to sample preparation procedures, LC separation conditions, and MS signal-to-noise optimizations at very low levels [12]. Several studies have been performed with various combinations of parameters and protocols for each of the three steps to seek improvements in the sensitivity, accuracy, efficiency, and cost of the T assays [5-6,10-12]. Sample preparation procedures are critical to achieving the success criteria, as they isolate, concentrate, and separate the analyte of interest from the biological sample into an LC-MS compatible matrix. This sample preparation workflow is primarily governed by the sample volume added for testosterone quantification, the method of testosterone extraction (different solutions used for protein precipitation, and the impact of pH on pellet formation), method of plating (semi-automated vs. manual), and sample reconstitution solvent. Additionally, the ease of automation increases the reproducibility and high throughput ability of the assay pertaining to robust clinical investigations. It is a summary of various sample preparation protocols for LC-MS/MS testosterone detection shown in Table 1. It showcases the unified approach to the selection of organic liquids, the volume ratios, ideal pH, as well as the different optimizations of the time of incubation and temperature conditions to primarily improve analyte extraction efficiency from other studies.
| Steps | Fariha et al. [11] | Sun et al. [8] | Wang et al. [12] | Hakkinen et al. [7] | Schofield et al. [1] | Thienpont et al. [4] |
|---|---|---|---|---|---|---|
| 1 | Sample Conditioning Sample (V=100 μL)+Acetonitrile (with 0.1% Formic Acid) mixed with Internal Standard (V=200 μL) | Sample Conditioning Sample (V=100 μL)+Calibration Liquid (V=100 μL) 15min mix+0.5 mol/L Ammonium Acetate (V=100 μL) pH=5.5 120 min mix RT | Sample Conditioning Sample (V=200 μL)+Acetonitrile (mixed with internal standards) (V=400 μL) | Sample Conditioning Sample (V=150 μL)+Calibration Liquid (V=20 μL) 15 min mix | Sample Conditioning Sample (V=500 μL)+Calibration Liquid (V=50 μL) | Sample Conditioning Sample (V=200 μL)+Calibration Liquid (V=20 μL) |
| 2 | Protein Precipitation Shake=10 min at 25°C Centrifuge=20 min at 4°C Decantation V=150 μL | LLE extraction nHexane+ethylacetate V=200 μL+300 μL 2× | LLE extraction Vortex=10 min Freeze at -20°C 10 min Vortex=10 min Centrifuge 10 min | LLE extraction Toluene V=1000 μL 10 min | LLE extraction hexane: ethyl acetate (9:1) V=2500 μL Centrifuge=2 min Freeze at -80°C 15 min | LLE extraction Methyl t-Butyl Ether V=1000 μL |
| 3 | Evaporation organic phase T=~60°C | Evaporation organic phase | N/A | Evaporation organic phase | Evaporation organic phase T=35°C | Evaporation organic phase T=50°C |
| 4 | Reconstitution 100 μL 70:30 (v/v) Acetonitrile: Water (with 0.1% Formic acid) | Dissolve Sodium Carbonate V=200 μL c=0.2mol/L pH=9.8 | Dissolve 200 μL supernatant+water (1:1) V=200 μL | Dissolve 30% Acetonitrile V=50 μL | Dissolve Methanol: Water (1:1) V=150 μL | Dissolve 0.7 mol/L Hydroxylamine in 3:7 Methanol/water V=50 μL T=70C 15 min |
| 5 | N/A | LLE extraction n Hexane V=500 μL 2× | N/A | N/A | N/A | N/A |
| 6 | N/A | Evaporation | N/A | N/A | N/A | N/A |
| 7 | N/A | Dissolve Methanol V=100 μL | N/A | N/A | N/A | N/A |
| 8 | LC-MS | LC-MS | LC-MS | LC-MS | LC-MS | LC-MS |
| Results | LOD=5 ng/dl Linear Range=5-1500 ng/dl CV<5% Accuracy ~93%-108% | LOD=0.5ng/dl Linear Range=1-1000 ng/dl CV<3.5% Accuracy=94%-108% | LOD=1ng/dl Linear Range=1-2000 ng/dl CV<3% Accuracy ~93%-104% | LOD=0.4ng/dl Linear Range=0.4-384 ng/dl CV<8% Accuracy=81%-98% | LOD=1 ng/dl Linear Range=1-1170 ng/dl CV<7% Accuracy ~97%-107% | LOD=0.5ng/dl Linear Range=1-2500 ng/dl CV<10% Accuracy ~89% |
Table 1: It is a summary of various sample preparation protocols for LC-MS/MS testosterone detection.
In this study, we discuss a sample preparation workflow that utilizes robotic liquid handlers for sample preparation automation, integrated with LC-MS/MS analysis for the quantification and monitoring of testosterone in human serum, a popular matrix in the clinical setting. The proposed sample preparation methodology takes into account a minute optimization of i) the choice of microplate used, based on plate area and sample volume ratio; ii) the time taken for sample incubation and phase-separation; iii) sample drying gas and temperature; iv) optimization of Multiple Reaction Mode (MRM) in mass spectrometry; v) method of plating; vi) choice of column and mobile phases and vii) LC ramping. Finally, based on the optimized method and workflow, a highly sensitive, reliable prototype kit for the precise measurement of testosterone in patient samples using LC-MS/ MS for clinical application was manufactured.
Chemicals and materials
Ultra-low steroid DDC Mass Spect Gold Serum® was purchased from Sigma-Aldrich and Testosterone (T-037 and T-070) in Acetonitrile were obtained from Cerilliant (RoundRock, TX, USA). Baker analyzed HPLC grade water, Acetonitrile (>99.9% purity), and Ethanol (>99.9% purity, gradient grade) were obtained from VWR. Formic Acid for LCMS was obtained from honeywell. Sodium Azide used was purchased from Sigma-Aldrich.
Quality control
To ensure the linearity series prepared for the assay functioned as expected, the National Institute of Standards and Technology (NIST) approved standard reference material 971a hormones in frozen human serum was used. SRM 971a (Male) was used in the development of the assay, with the testosterone measuring at 580.8 ± 9.0 ng/dL mass concentration. SMR 971a was stored at -80°C and thawed to room temperature when ready for testing. The internal standard for the assay was prepared by diluting the Cerilliant T-070 Testosterone (at 1 mg/ mL) (13C3C16H28O2 (MW=291.40)) two-hundred folds in HPLC grade ethanol (500 ng/mL). This solution was further diluted one-hundred folds in acetonitrile with 0.1% formic acid solution to prepare the Daily Working Solution (DWS).
Phase testing
The isotopically unlabeled testosterone was dissolved at 10 ng/mL concentration at varying ratios of HPLC water and acetonitrile at from 100% to 0% v/v ratio of the solvents and injected in the LC-MS/MS system for analysis. Figure 1 shows the Relative Intensity (RI) overlay plot from this test, that helped identify a Water/Acetonitrile=70/30 v/v ratio as the optimum condition for sample injection and analysis.

Figure 1: Relative Intensity (RI) overlay plot for different organic to inorganic solvent ratio used for T dissolution. Red line shows premature elution when T was dissolved in 100% acetonitrile. Green line shows a delay in Retention Time (RT) when T was dissolved in 100% water. Lines blue (Water/Acetonitrile=65/35) and indigo (Water/Acetonitrile=70/30) show the best chromatogram shapes with highest RI (~200000). Water/ Acetonitrile=70/30 ratio was chosen due to the lower signal-to-noise ratio.
Calibrator preparation
Sodium azide (0.05%) and thawed DDC Mass Spect Gold Serum® were used as the base for the calibrators. Two intermediate ‘bulk’ solutions were prepared using the isotopically unlabeled T-037 testosterone solution purchased from Cerilliant (at 1 mg/mL) (C19H28O2 (MW=288.42)). This solution was diluted to 500 ng/mL and 12.5 ng/mL in gradient grade ethanol to form Bulk Solution A and B, respectively. These bulk solutions were added to spike the gold serum base to form six-concentration calibrators via overnight rocking.
Sample preparation
All samples were brought to room temperature, and on a large volume conical bottom Axygen shallow 96-well plate, 100 μL of the calibrators were added to 200 μL of DWS. This step was investigated with deep well v-bottom 96-well plates, standard v-bottom 96-well PCR plates, as well as vial-based preparation techniques. The role of the well-depth and shape on the automation of the procedure is further discussed in the results sections. The DWS consisted of acetonitrile, with 0.1% formic acid, as well as an internal standard. The samples were shaken using the TriNEST™ microplate shaker at 700 RPM for ten minutes, and then centrifuged at 4600 RPM at 4°C for twenty minutes. The organic layer was decanted to a separate shallow conical bottom 96-well Nunc plate, evaporated to dryness at ~60°C with a constant airstream, and then reconstituted with 100 μL of HPLC water: Acetonitrile (70:30 v/v) in 0.1% formic acid solution. The plate used for decantation was also investigated with standard v-bottom 96-well PCR plate, as well as a vial-based method. Additionally, the reconstitution solvent was tested for the effect of pH, with no acid, 0.1% formic acid and 1% formic acid over 3-runs shown in Figure S1. The samples were given a final ten-minute shake at the TriNEST™ at 400 RPM before being injected to the LC-MS/MS.
Carryover
Carryover for the study was performed by first establishing baselines for the highest and lowest concentrations of the calibrators. Following running working solutions, the highest calibrator (single injection) was injected into the system immediately followed by injection of the lowest concentration of the calibrator (in triplicate). Any significant change in the measured concentration of testosterone was taken as an indication of carryover. The measured concentration of the lowest calibrator concentration was equated as a measure of sample carryover, running it in the same plate following the injection of the highest calibrator concentration, in triplicates for seven different sets.
LC-MS/MS equipment and conditions
The PerkinElmer QSight™ 200 MassSpec triple quad liquid chromatography system, together with the QSight™ LX-50 Pump, QSight™ LX-50 Oven and QSight™ LX-50 Autosampler, was used with an ESI source. HPLC column (3 μm, C18, 100 Å, 50 × 3.0 mm) with guard column (2.7 μm Brownlee SPP) were used with the system. The triple quad mass spec was operated with the nebulizer gas pressure at 200 psi, electrospray voltage of 5850 volts, and a source temperature of 425°C. The total run time for each sample was 3 minutes, with 429 scans, with the system being operated in Multiple Reaction Monitoring (MRM) mode. Analytes were resolved by reversephase HPLC prior to mass spectrometry analysis to reduce the level of interfering compounds and provided a characteristic chromatograph with retention time for each analyte. The first quadrupole (Q1) was set to filter out all ions except for the precursor (analyte or internal standard). Q3 filtered out all but one selected testosterone ion, thus a signal was only detected when a selected precursor generated a specific fragment (MRM transition). Analyte and internal standards specific to MRMs were monitored sequentially during chromatographic separation. Signals were quantitated only if the specified MRM was recorded at the correct analyte retention time. Thus, quantitation in this MS/MS method is very specific. ‘Quantifier’ corresponds to testosterone with Q3 mass=97 amu, and ‘Qualifier’ corresponds to testosterone with Q3 mass=109 amu and will be referred to as such throughout the paper. For the method developed, the retention time for each sample was 1.84 minutes (with the matrix). The LC method for this study was established such that there was a fast full-loop flush at 2.00 mm needle height from the bottom of the well at 14 μL flush volume. With HPLC water as the weak solvent (Mobile Phase A) and LC-grade acetonitrile as the strong solvent (Mobile Phase B), the analytes were loaded at a flow rate of 0.600 mL/min flow rate initially. It illustrates the optimized MRM parameters, and the optimized conditions for the LC method applied in this study, respectively shown in (Tables 2a and 2b). The set point temperature for the samples was maintained at a steady 4°C. The column was maintained at a steady temperature of 40°C for the duration of the analysis. The data was collected using the Simplicity 3Q software made for QSight, version 2016-2019.
| Analyte | Type | Q1 mass | Q3 mass | RT (min) | EV | CC | CCL2 |
|---|---|---|---|---|---|---|---|
| Testosterone | Quant | 289.2 | 97 | 1.84 | 20 | -34 | -56 |
| Quant IS | 292 | 100 | 1.84 | ||||
| Qual | 289.2 | 109 | 1.84 | 20 | -37 | -64 | |
| Qual IS | 292 | 112 | 1.84 |
Note: RT refers to the retention time (in minutes).
Table (2a): Mass parameter table for T (quantifier and qualifier) and their respective internal standards.
| Time (min) | Flow rate (mL/min) | %A | %B |
|---|---|---|---|
| 0 | 0.6 | 70 | 30 |
| 1.00 | 0.6 | 70 | 30 |
| 2.00 | 0.6 | 50 | 50 |
| 2.10 | 0.6 | 5 | 95 |
| 2.50 | 0.6 | 5 | 95 |
| 2.51 | 0.6 | 70 | 30 |
| 3.00 | 0.6 | 70 | 30 |
Table (2b): Solvent flow rate for weak and strong solvents used in the mobile phases for the LC-MS/MS extraction and analysis of testosterone.
Automation
To further adapt the testosterone prototype kit for a robust research application, a semi-automated sample preparation method was created using the JANUS® G3 liquid handler (PerkinElmer Corp.,). The sample preparation process previously described was adapted onto the JANUS platform using the Janus Application Assistant and WinPREP® for Janus® software. Figure 2a showcases the finalized deck layout of the sample preparation protocol, while Figure 2b shows the on-deck and off-deck steps, color-coded by human interaction. Owing to the complexity of some of the steps in the sample preparation process (albeit as simple as protein preparation), the plate had to be taken off-deck several times for shaking, centrifugation and drying, thus the use of the “semi-automated” terminology. Finally, to determine the efficacy of this automated sample preparation method, manually plated samples were compared via LC/MS analysis against automated plates with regards to precision as well as preparation speed.

Figure 2: (a) Automated Sample Preparation Deck Layout; (b) Sample preparation automation workflow analyzed by time for both on and off deck protocol steps. Blue boxes represent the on-deck steps while grey boxes represent the off-deck steps. Times staggered above and below the workflow indicate times in Minutes; Seconds; Milliseconds measured with a stopwatch.
For all the studies, statistical significance was established using a student’s t-test, where p-value<0.05 was considered statistically significant between averaged concentrations measured. For the carryover study, the result was investigated further using a boxplot diagram to identify any outliers (none identified), and for the accelerated stability study, the normalized concentrations were compared by generating a comparative letter report using Tukey’s HSD post-hoc test. All statistical analysis was performed using JMP 15 Pro software.
Method validation
At the beginning of the study, it was important to confirm that the manufactured calibrators were indeed giving the expected readouts before any further studies could be performed. Therefore, the calibrators were plated in sextuplicate for quantification and validated against the NIST material. Figure S2 shows the average concentrations measured for each calibrator level and the NIST material alongside the known concentration. The NIST material measured very close to the theoretical value (5.80 ± 0.01), and the same was observed for the calibrators prepared, even for the lowest concentration of 0.05 ng/ mL. Once the concentrations were established, the calibrators were then used as ‘standards’ for the other experiments, moving forward. Upon multiple studies, it was established that the Axygen 96-well conical bottom plates were ideal for the initial plating, and the larger surface area of the Nunc 96-well plate made it ideal for the drying and reconstitution step (data for two of the plates used are reported in Figure S3, along with Table TS1 outlining the difference in preparation methodologies).
Precision, linearity and accuracy
Linearity, accuracy, and precision were performed over a 6-day study, where each calibrator level was plated in triplicates, and the data was obtained from a 6-point calibration curve using the Simplicity 3Q software. Calibrator analyte/IS ratios were plotted (y-axis) against the calibrator concentrations (x-axis), and a standard curve was generated using linear regression with 1/x weighting. Standard curve fitting via linear regression was also performed on Simplicity 3Q, which was then utilized to determine both inter-assay linearity as well as sample accuracy, allowing for the measurement of individual analyte Coefficient of Variation (CV). Total precision was calculated as %CV for each level within run, between run, and for all 6 runs. (Table 3) shows the average concentration for the calibrators, the %CV and % of Accuracy for quantifiers (top) and qualifiers (bottom) for the full study. For each plate, quality control samples (low=2 ng/mL; high=10 ng/mL) were run together with the calibrators for method validation. These quality control samples were prepared with the calibrators from the same standard reference material lot and tested with the NIST material. The R-square values were taken as a measure of the linearity based on a standard linear regression curve. Figure 3 illustrates the observed linear trend (for automated and manual sample preparation), where R-Square values measure at 0.99 and above for both runs. Sample accuracy was similarly obtained from the simplicity 3Q software and averaged for all the plates across the study days.
| Calibrator level | μx ± σ (in ng/mL) | %CV | % Accuracy |
|---|---|---|---|
| L1 | 0.05 ± 0.005 | 5.6 | 90.6 |
| L2 | 0.28 ± 0.005 | 3.7 | 107.3 |
| L3 | 2.00 ± 0.060 | 2.1 | 97.5 |
| L4 | 5.32 ± 0.060 | 1.7 | 106.9 |
| L5 | 9.77 ± 0.119 | 1.9 | 99.5 |
| L6 | 14.67 ± 0.227 | 1.4 | 98.3 |
| Calibrator level | μx ± σ (in ng/mL) | %CV | % Accuracy |
| L1 | 0.05 ± 0.003 | 6.1 | 89.1 |
| L2 | 0.29 ± 0.020 | 5.5 | 109.4 |
| L3 | 1.98 ± 0.052 | 2.5 | 96.7 |
| L4 | 5.39 ± 0.080 | 1.7 | 107.3 |
| L5 | 9.83 ± 0.129 | 1.8 | 99.3 |
| L6 | 14.67 ± 0.283 | 1.7 | 98.3 |
Note: (Top: Quantifier; Bottom: Qualifier).
Table 3: %CV was used as a measure for precision, determined using 3 replicates per level each day, over a 6-day period (n=18 per level). Samples were found to be within 6.2% or less for intraday and interday measurements, inclusive of both MRMs. The average % accuracy for all calibrators was found to be 100 ± 20, inclusive of both MRMs as well. These values fall well within our ≤ 20% cut-off value.

Figure 3: Standard curve comparison, to illustrate performance differences between manual vs automated plating of calibrators L1-L6 in triplicates (n=3 per calibrator per plate). Both plates were run on the same day, with the results showcasing comparable performance.
Accelerated stability
The stability of the calibrators was tested via an accelerated stability study where the samples were subjected to extreme temperature changes (from -20°C storage to 30°C) at two-day intervals, over a period of 14 days. Using the equation:
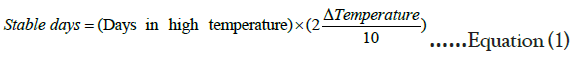
Equation 1. Modified Arrhenius’ Equation
We were able to identify how long the calibrators would be stable without any degradation. This was important in the design process of a prototype kit that can be used in clinical settings.
Duplicates of calibrators were transferred from -20°C storage to 30°C every two days, and on the 14th day, samples were harvested. All the samples were then tested against fresh (Day 0) samples. Samples were mainly stable over the 14 days, and from the stability approximation obtained from equation 1, the calibrators are expected to be stable for up to 488 days as shown in Figure S4. Calibrator 1 had no specific trend, and it was also only slightly above the background. Therefore, by virtue of the trends observed for all other calibrators, it was assumed that Calibrator 1 had the same trend.
Freeze-thaw
Once results were obtained for precision, linearity, and accuracy, prototype kits were produced using the calibrators (six concentrations of testosterone, two quality control calibrators, and an internal standard vial), that were stored at -20°C. The prototype kit was subjected to six freeze-thaw cycles (between -20°C and room temperature), and changes in the measured concentration of testosterone calibrators over the freeze-thaw cycles after thawing to room temperature in triplicate were obtained in (Figure 4). No significant degradation was observed in any of the measured testosterone values over the six freeze-thaw cycles.

Figure 4: Average testosterone concentration for quantifier and qualifier for different calibrator levels and quality control (low and high) samples. No significant difference was observed in the measured value of testosterone for all the different calibrators over six freeze-thaw cycles.
Automation
To assess the proposed prototype kit’s adaptability for large-scale sample testing, the developed semi-automated sample preparation process was tested against manual plating. Overall, automation efficacy was directly observed by comparing the necessary plating time, as well as overall plate performance (linearity, precision, and accuracy with regards to the calibrators plated in triplicate against a manually prepared plate). The semi-automated protocol yielded an overall runtime of 27 minutes 16 seconds (including off-deck actions), compared to the manual plating time of 17 minutes, obtained over an average of 6 runs. As mentioned previously, the automated protocol’s runtime is increased due to the complexity of serum as a matrix, which required additional air gaps to be created within each pipetting tip used. This required the protocol to be programmed with 10 μL of air aspirated pre-aspiration and post-dispense of the calibrators and samples, before discarding the tip. This additional step was part of both the initial calibrator and sample pipetting step, as well as post-centrifugation supernatant separation step. For the first step, it was observed that human serum, when shaken (resulting from sample transportation and handling) or vortexed (for calibrators and QCs, once thawed to room temperature and prior to pipetting) almost always formed a thin film towards the opening of the container, which the robotic liquid handler’s sensing technology would often misinterpret as the starting liquid height. Hence, this issue was mitigated by enabling the ‘Ultra’ sensing option for the liquid search height instead of ‘High’ sensing option with a liquid height verification feature, in addition to the air aspirations. For the latter step, i.e., supernatant separation, the viscosity and volatility of the serum was impacted due to the addition of the organic solvent present in DWS, which resulted in significant sample dripping a critical challenge observed when using multichannel pipettes for manual preparation as well. Adding the air aspiration step provided an easy solution to this problem, which is not possible to apply for manual preparation. For the other steps, i.e., adding the DWS and the reconstitution solvent, significant time was added due to the prewetting steps. Both aforementioned solvents were composed of volatile liquids leading to significant dripping, which was mitigated using 3 cycles of pre-wetting the tips, with 150% of the aspirating volume. This not only prevented the dripping, but also ensured accurate volume dispenses per individual sample, even with multiple tips in action. Additionally, some of the off-deck tasks in the protocol included sample shaking, centrifugation, as well as drying, which are not readily available features for conventional liquid handlers, and upgrades to include them would make the process expensive. The performance of the automated plating was similar to that of manual plating, with a reported R-Square value for the observed linearity of R2=0.9994 for the automated plate, and R2=0.9962 for the manual plate, with no statistically significant difference shown in Figure 3. The high linearity of the standard curves showcases both the proposed assay’s linearity over the required range of testosterone concentrations, as well as the automatability and the high-throughput capacity of this kit and increasing applicability of its utilization in clinical settings with patient samples.
Herein, this study successfully establishes a chromatographic assay with LC-MS/MS analysis for testosterone detection in serum using a prototype kit with automation incorporation for clinical adaptability. This can play a pivotal role in screening for hormonal disorders, as well as for research purposes when developing treatments for said hormonal disorders. Overall, the improvements this study proposes for testosterone is of tremendous value to the scientific community for better understanding of a rather simplistic workflow for sample preparation.
We would like to thank Joe Trometer and Cel Welch for their valuable insight during the initial phase of this study. A.T. is a paid scientific advisor/consultant and lecturer for PerkinElmer.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Fariha R, Hill C, Jabrah M, Spooner A, Tripathi A (2024). Automated Preparation of Serum Testosterone for Detection Using LC-MS/MS. Andrology. 13:329.
Received: 12-Aug-2024, Manuscript No. ANO-24-33473; Editor assigned: 15-Aug-2024, Pre QC No. ANO-24-33473 (PQ); Reviewed: 29-Aug-2024, QC No. ANO-24-33473; Revised: 05-Sep-2024, Manuscript No. ANO-24-33473 (R); Published: 12-Sep-2024 , DOI: 10.35248/2167-0250.24.13.329
Copyright: © 2024 Fariha R, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.