Mycobacterial Diseases
Open Access
ISSN: 2161-1068
ISSN: 2161-1068
Research Article - (2013) Volume 3, Issue 2
Keywords: Mycobacterium tuberculosis; Potential drug; REACTOME database; Anti-tubercular compounds
Despite the wide variety of anti-tuberculosis drugs, tuberculosis (TB), caused by Mycobacterium tuberculosis (MTB), remains a public health challenge today, claiming millions of lives and new cases every year. This success of MTB is in part due to the discontinuity of its life cycle in the host system, owing to its ability to enter and exit from different states in response to the antimycobacterial host defense mechanisms, enabling it to infect, grow, persist and survive in human macrophages [1]. Enhanced service provision of the existing antibiotics in recent years through Direct Observed Treatment (DOT) as implemented by the World Health Organization (WHO) is of immense value in controlling the disease. Unfortunately, these drugs have several shortcomings, the most important being the emergence of drug resistance, making even the front-line drugs ineffective. In addition, the deadly interaction between TB and Human Immunodeficiency Virus (HIV)/Acquired Immunodeficiency Syndrome (AIDS) is threatening to compromise gains in TB control, leading to further challenges for anti-tubercular drug discovery [2].
TB is currently treated with a decades-old drug regimen, lasting at least six months [3,4] using the initial combination of isoniazid, rifampin, pyrazinamide, and ethambutal as front-line drugs [5], a control strategy implemented by WHO in response to the global TB epidemic. Unfortunately, there is no guarantee of the complete sterilization of the infection and non-compliance with this long duration of TB treatment contributes to the development of resistance. In addition, the global HIV/AIDS epidemic has led to an explosive increase in TB incidence and contributes to increases in multidrugresistant TB (MDR-TB) prevalence [6], which is a form of TB that is resistant to at least two of the most commonly used drugs in the current four-drug or first-line therapies (isoniazid and rifampin). As current anti-tuberculosis drugs are not sufficiently efficient, prone to development of multi-drug resistance and no new anti-tuberculosis drugs have been designed for over 20 years, it is increasingly important to pursue new and effective strategies to confront the challenge of TB in this 21st century.
In view of TB challenges, the goal of eradicating TB in the coming years depends on the development of new diagnostics, treatment and prevention tools, including drugs and vaccines [1]. Any new effective drug should be able to shorten the duration of the treatment, improve the treatment of MDR-TB, and possibly provide an effective treatment of latent TB infection. Thus, in addition to being compatible with Antiretroviral Drugs (ARVs) used to stop the progression of HIV disease, the properties of anti-TB agents must include antibacterial activity, capacity to inhibit the development of resistance, and ability to kill the intracellular organisms that are in a persistent state. For this, target identification and validation are essential for the success of the drug discovery and development process. However, the identification of novel drug targets for diseases and development of new drugs have always been expensive and time-consuming and the amount of time required for designing a new drug is still high, approximately ten to fifteen years [7].
Currently, technological developments in large-scale biological experiments, coupled with bioinformatics tools, can provide inexpensive strategies which shorten the length of time spent in drug discovery. This can be achieved through integration of functional and genomics data to generate the organism under consideration’s proteinprotein functional interaction networks [8-10]. This approach provides the op opportunity to look at genes within their context in the cell for the global analysis of whole genomes, allowing the identification on of key proteins essential to the pathogen’s growth, survival and viability using network topological properties. Another benefit of this model is that it enables the expansion of the range of potential drug targets and leads to optimal target-based strategies, free of trial and error. We applied this strategy to MTB [10] and 881 proteins were found to be critical in maintaining the system’s integrity, influencing the system ‘s robustness and stability under perturbations, thus helping the bacterial pathogen to survive and achieve its goal within the host. Filtering this list of key proteins can produce appropriate and effective new drug targets for the development of novel drugs with new biological mechanisms of action against drug susceptible and drug-resistant strains.
The identification of drug targets requires consideration of a variety of criteria, including understanding the complex interplay between pathogen and host, as these interactions are key factors in determining the outcome of the MTB infection. Considering the host system could yield more suitable drug targets that prevent potential adverse reactions in the host. Here, we use human-MTB functional interactions predicted by integrating functional and genomics data to filter the list of 881 potential targets for MTB by considering only proteins or genes which do not interact with human proteins unless they have different functions, as potential drug target candidates.
Previously generated human and MTB intra-species functional networks were used. These functional networks were constructed by combining protein interaction data from the STRING database [11,12] and complemented by additional interaction data from sequence and microarray data for the MTB network [10,13] and by Bossi and Lehner’s interaction data [14], together with data from the REACTOME database [15] for the human network, as depicted in Figure 1. For the human-MTB inter-species functional network, we are using the functional interactions previously retrieved through manual curation of the literature and predicted using the interologs method and filtered using gene expression data [16]. As in the case of the MTB functional network, these inter-species functional interactions are complemented by functional interactions from sequence data, more precisely interactions predicted from protein sequence similarity and shared domains between proteins from the InterPro database (http:// www.ebi.ac.uk/interpro).

Figure 1: Flowchart depicting MTB protein target filtering. The chart describes the construction of Human, MTB and Human-MTB functional interaction networks and use of these networks to filter the list of MTB protein targets identified from the MTB network.
Building unified human and MTB intra-species functional networks
Interaction data used to build intra-species human and MTB functional interaction networks were retrieved from diverse sources. The uncertainty of data and noise inherent in each source were managed by systematically weighing or scoring these functional associations [9]. These scoring schemes are data source and technology dependent i.e., a given scoring scheme varies according to the data sources and is designed on the basis of the technology used. Functional interactions from the STRING database were used with confidence scores as defined by the STRING scoring schemes.
For each evidence source, functional interaction scores were categorized into three different confidence levels, namely low, medium and high confidence. All interactions whose scores are strictly less than 0.3 were considered to be low confidence, scores ranging from 0.3 to 0.7 (0.3 ≤ score ≤ 0.7) were classified as medium confidence and scores greater than 0.7 yield high confidence interactions. The final combined score was computed by combining all confidence scores between two proteins i and for all datasets through a unified network, under the assumption of independence and given by
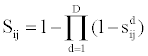 (1)
(1)
where 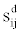 is the confidence score of a functional interaction between i and j predicted using the type of data d, and D the number of interaction sources in the dataset under consideration.
is the confidence score of a functional interaction between i and j predicted using the type of data d, and D the number of interaction sources in the dataset under consideration.
From a unified functional network, a reliability threshold was applied to reduce the impact of bias in functional interactions coming from experimental predictions and computational approaches [10]. For intra-species functional networks we only considered interactions whose confidence scores ranged from medium to high confidence and for functional interactions with low confidence, only those predicted by at least two different approaches were considered, in order to produce a network of reasonable confidence interactions and coverage. This produces a human functional network with 17847 proteins and 710142 interactions and a final MTB functional network containing 4136 proteins and 59919 interactions. The MTB functional network was used to identify the system’s key proteins using network topological properties and 881 proteins were predicted to be potential MTB drug targets [10].
Building a unified Human-MTB inter-species functional network
As pointed out previously, we are using functional interaction pairs retrieved through manual curation of the literature and predicted using the interologs method. Interologs are interacting proteins in one organism whose corresponding orthologs are also predicted to interact in another organism [17]. For predicting these interologs, experimentally verified interactions between human and bacterial proteins were extracted from the Pathosystems Resource Integration Center (PATRIC) [18] and the Host-Pathogen Interaction database (HPIDB) [19], and intra-species interacting pairs of proteins from the Database of Interacting Proteins (DIP) [20] were also collected. For each interaction, orthologs of both of the proteins were identified in human and MTB proteomes, respectively, and we inferred that these orthologs also interact. Ortholog files were downloaded from the Integr8 project [21] (http://www.ebi.ac.uk/integr8) at the European Institute of Bioinformatics (EBI). These interactions are complemented by additional interaction data from protein sequence similarity and conserved protein domains scored using information theoretic-based approaches described in [9]. Here, we performed the Basic Local Alignment Search Tool (BLAST) algorithm [22,23] for sequence similarity searching, specifically we ran the blast all program, aligning all MTB protein sequences against the human protein sequences and vice versa using same parameters as in [9].
Functional and statistical analysis
To gain insight into the biological processes of the proteins involved in human-MTB interactions, we functionally com-pared predicted human-MTB protein pairs to random protein pairs by computing their Gene Ontology (GO) biological process similarity. We used the GO-universal metric [24] based on the GO-DAG topology [25,26] to compute the biological similarity between two proteins. This also allows us to assess the relevance of this predicted human- MTB functional inter-actions as functional interactions are likely to occur between proteins involved in similar biological processes [27]. Thus, for each pair of proteins involved in a predicted human-MTB functional interaction, we computed the semantic similarity between their GO annotations and compared these similarity scores on average to scores between interacting proteins from random human-MTB functional interactions.
Deriving statistically significant GO annotations among these inter-species interacting proteins is based on the hyper-geometric distribution model. This model consists of computing a p-value for each term using its frequencies of occurrence in the experiment. This p-value is the probability that the number of genes or proteins annotated with the term under consideration in the target set occurs by chance or is comprised of randomly drawn genes from the reference or background set. We used the Bonferonni p-value, which is a corrected p-value for multiple testing, and we selected those GO terms enriched in our target protein list by requiring a p-value less than 0.05. The human and MTB protein annotation data were downloaded from the Gene Ontology Annotation (GOA) project [28] (http://www.ebi.ac.uk/ goa).
We used human, MTB intra-species functional networks and a human-MTB inter-species functional network to filter a list of 881 protein targets previously identified within MT B using topological properties of its protein-protein functional interaction network [10]. To achieve this, we overlaid predicted human-MTB functional interactions onto the human and MTB (strain CDC1551) proteinprotein functional interaction networks to analyze inter-species interacting proteins and to uncover suitable protein targets.
Comparing functional similarity scores between predicted and random Human-MTB interacting proteins
A GO semantic similarity measure can be used to assess the functional similarity between protein pairs of predicted functional interactions [27]. We used 4616 functional relation-ships predicted between 1011 human and 626 MTB proteins to determine whether functional similarity scores between these interacting proteins tend to be significantly different from those obtained after generating random interactions between human and MTB proteins. Using the Biological Process (BP) ontology, we obtained an average similarity score of 0.31 between predicted interacting human-MTB proteins and of 0.06 for random interactions between human and MTB proteins.
To determine whether this difference was statistically significant and not merely due to chance, we estimated the distribution of average functional similarity scores for random interactions using a Monte Carlo sampling procedure (Figure 2). When compared to this distribution, a nonparametric p-value <2.2e-16 was obtained under the hypothesis that the central measure score of randomly drawn interactions is less than that of predicted human-MTB interactions. This indicates that functional similarity scores of the predicted interactions are significantly higher than random interactions, suggesting that the predicted interactions tend to be involved in similar biological processes, and thus are plausible [27,28].

Figure 2: Analyzing human-MTB interacting protein functional similarity scores: Predicted human-MTB functional interactions lead to significantly higher functional similarity scores compared to random human-MTB interactions. The estimated distribution of random human-MTB interacting protein functional average similarity scores is used to determine whether the predicted human-MTB interacting protein functional similarity average score (μ=0.31) is significantly high. The red line indicates the approximate probability density function of the distribution of random human-MTB interacting protein average functional similarity scores, estimated using a Gaussian smoothing kernel.
Clustering interacting proteins and enrichment analysis
We used Human-MTB interacting proteins to build weighted networks from human and MTB functional networks. We generated a human weighted network of 889 out of 1011 proteins predicted to interact with MTB proteins intra-connected by 9042 interactions and an MTB weighted network of 598 out of 626 proteins predicted to interact with human proteins intra-connected by 10574 interactions. Note that these networks are weighted using protein functional similarity scores computed using the GO-universal metric. Thereafter, we clustered proteins from each organism in different classes or communities [29] and performed enrichment analysis to highlight the most relevant GO terms associated with a given gene list in each cluster compared to all annotated genes in the network. 11 clusters have been identified for human proteins predicted to interact with MTB proteins and 10 for MTB proteins predicted to interact with human proteins. Results are shown in Table 1 for human and Table 2 for MTB with the two most statistically relevant terms for each cluster.
| Cluster | Proteins | Enriched Terms | GO Ids | GO term | Level | p-value | Bonferonni-correction |
|---|---|---|---|---|---|---|---|
| 1 | 135 | 18 | GO:0043066 | negative regulation of apoptotic process | 7 | 6.99253×10−09 | 3.41235×10−06 |
| GO:0006987 | activation of signaling protein activityinvolved in unfolded protein response | 11 | 1.80733×10−08 | 8.81979×10−06 | |||
| 2 | 121 | 19 | GO:0010467 | gene expression | 4 | 0.00000 | 0.00000 |
| GO:0006120 | mitochondrial electron transport, NADHto ubiquinone | 9 | 1.37596×10−10 | 4.93969×10−08 | |||
| 3 | 95 | 29 | GO:0044281 | small molecule metabolic process | 3 | 0.00000 | 0.00000 |
| GO:0006006 | glucose metabolic process | 7 | 2.69909×10−10 | 1.08233×10−07 | |||
| 4 | 152 | 56 | GO:0042738 | exogenous drug catabolic process | 10 | 0.00000 | 0.00000 |
| GO:0071236 | cellular response to antibiotic | 6 | 3.68974×10−05 | 0.02007 | |||
| 5 | 77 | 25 | GO:0044255 | cellular lipid metabolic process | 4 | 0.00000 | 0.00000 |
| GO:0033540 | fatty acid beta-oxidation usingacyl-CoA oxidase | 11 | 0.00000 | 0.00000 | |||
| 6 | 105 | 38 | GO:0034641 | cellular nitrogen compound metabolicprocess | 3 | 0.00000 | 0.00000 |
| GO:0002376 | immune system process | 1 | 1.25410×10−05 | 0.00515 | |||
| 7 | 50 | 16 | GO:0006183 | GTP biosynthetic process | 11 | 0.00000 | 0.00000 |
| GO:0006283 | transcription-coupled nucleotide-excision repair | 9 | 2.02819×10−08 | 4.94879×10−06 | |||
| 8 | 112 | 26 | GO:0034220 | ion transmembrane transport | 6 | 1.95507×10−10 | 1.302076×10−07 |
| GO:0008543 | fibroblast growth factor receptorsignaling pathway | 8 | 1.89730×10−09 | 1.26360×10−06 | |||
| 9 | 39 | 9 | GO:0006457 | protein folding | 6 | 0.00000 | 0.00000 |
| GO:0000398 | mRNA splicing, via spliceosome | 11 | 3.49331×10−11 | 4.89063×10−09 | |||
| 10 | 1 | 2 | GO:0006950 | response to stress | 2 | 0.00590 | 0.01179 |
| GO:0006457 | protein folding | 6 | 0.01383 | 0.02765 | |||
| 11 | 2 | 12 | GO:0050796 | regulation of insulin secretion | 8 | 7.81617×10−06 | 0.00016 |
| GO:0006112 | energy reserve metabolic process | 5 | 1.25409×10−05 | 0.00025 |
Table 1: Clustering human proteins predicted to interact with MTB proteins. Different clusters identified and the two most statistically relevant GO biological process terms associated with a given gene list in each cluster.
| Cluster | Proteins | Enriched Terms | GO Ids | GO term | Level | p-value | Bonferonni-correction |
|---|---|---|---|---|---|---|---|
| 1 | 107 | 6 | GO:0006418 | tRNA aminoacylation for proteintranslation | 10 | 4.73638×10−11 | 7.72030×10−09 |
| GO:0006164 | purine nucleotide biosynthetic process | 9 | 4.07330×10−06 | 0.00066 | |||
| 2 | 45 | 8 | GO:0033216 | ferric iron import | 11 | 0.00035 | 0.02514 |
| GO:0052099 | acquisition by symbiont of nutrientsfrom host via siderophores | 6 | 0.00035 | 0.02514 | |||
| 3 | 84 | 8 | GO:0006099 | tricarboxylic acid cycle | 8 | 0.00000 | 0.00000 |
| GO:0006096 | glycolysis | 9 | 2.26606×10−06 | 0.00027 | |||
| 4 | 67 | 6 | GO:0006825 | copper ion transport | 9 | 5.7285×10−07 | 3.72353×10−05 |
| GO:0015986 | ATP synthesis coupled proton transport | 12 | 7.95668×10−07 | 5.17184×10−05 | |||
| 5 | 107 | 6 | GO:0055114 | oxidation-reduction process | 3 | 0.00000 | 0.00000 |
| GO:0006631 | fatty acid metabolic process | 8 | 0.00042 | 0.03869 | |||
| 6 | 121 | 8 | GO:0040007 | growth | 1 | 0.00000 | 0.00000 |
| GO:0006457 | protein folding | 6 | 2.89594×10−08 | 4.72038×10−06 | |||
| 7 | 64 | 3 | GO:0055114 | oxidation-reduction process | 3 | 1.39651×10−06 | 5.16710×10−05 |
| GO:0019367 | fatty acid elongation, saturatedfatty acid | 11 | 1.85197×10−05 | 0.00069 | |||
| 8 | 1 | 0 | - | - | - | - | - |
| 9 | 1 | 0 | - | - | - | - | - |
| 10 | 1 | 1 | GO:0006508 | proteolysis | 5 | 0.023509 | 0.02351 |
Table 2: Clustering MTB proteins predicted to interact with human proteins. Different clusters identified and the two most statistically relevant GO biological process terms associated with a given gene list in each cluster.
The human proteins predicted to interact with MTB proteins were specifically enriched in recognizing the presence of the infection and are fundamentally involved in responding to the MTB infection. For example, certain human proteins, especially those belonging to cluster 1, are enriched in processes related to negative regulation of apoptosis, which is in agreement with the fact that virulent MTB strains inhibit apoptosis of the host macrophage at the early infection stage to protect their replicative niche [30] by up-regulating the NF-κB signaling pathway, resulting in the up-regulation of FLIP, an inhibitor of death receptor signaling [31]. These human proteins could enable the bacterial pathogen to survive and persist in the macrophages for a long time.
The persistence of MTB in the host system depends on the ability of the bacterial pathogen to acquire and utilize nutrients from the interior of the macrophage phagosome. MTB proteins predicted to interact with human proteins are particularly enriched in biological processes that allow the bacterial pathogen to acquire nutrients from its host (Table 2: cluster 2). In particular, MTB imports iron, which is an indispensable nutrient for almost all organisms [32] and is essential for growth of MTB [33]. It is known that after its establishment in a specific environment, MTB switches its metabolic pathways to utilize fatty acids rather than carbohydrates and as shown in Table 2, several clusters contain proteins enriched in fatty acid metabolism. This suggests that the predicted human-MTB interacting proteins are crucial for MTB survival, intracellular lifestyle and spreading strategies. These proteins provide insight into how MTB might acquire nutrients and how it modulates the host response to its advantage. They may play a role in protecting the pathogen from the environment through interaction with host proteins.
Predicted interactions and drug targets
A total of 9707 interactions were predicted between 2259 human and 633 MTB proteins. Among these 2259 human proteins, only 1011 are found in the human functional network generated and 626 MTB proteins out of 633 are found in the MTB functional network. These human and MTB proteins are connected by 4616 interactions out of the total of 9707 predicted interactions. These inter-species interactions are used to overlay the human network onto the MTB network and identify MTB protein targets (of the 881 targets) predicted to interact with human proteins.
We used the hyper geometric test to determine whether the predicted list of proteins contains more drug targets than expected by chance. Among 626 MTB proteins predicted to interact with human proteins, 275 are in the drug target list.
The statistical test checks whether 275 targets out of 626 MTB proteins predicted to interact with human proteins is more than random chance compared to the background of 881 MTB potential drug targets out of 4136 proteins contained in the MTB functional network. Performing the hyper geometric test, a p-value of 3.773454e -45 was obtained showing evidence that the list of MTB proteins interacting with human proteins contains more drug targets than would be expected by chance.
We then used different criteria to filter the 275 interacting proteins, as well as non-interacting drug targets to identify the most suitable targets using the pipeline described in Figure 3. For the 275 targets that interact with human proteins, 259 were predicted to interact based on sequence similarity and shared domains, and only 16 targets were predicted to interact with human proteins through interologs. The 259 MTB drug targets predicted to interact with human proteins should be neglected as potential candidates for further target based drug development. This is because they may adversely impact the host system or can lead to unwanted toxicity as they have similar sequences and functions to their human partners. We checked the rest for paralogs, using paralog candidates from the Ensembl database [34] (http://www.ensembl.org/) and mapping Ensembl IDs to UniProt IDs. Of the remaining 16 proteins that interact with human proteins, only 2, DnaB and RecN, were found to have no paralogs. DnaB is essential for growth [35,36], and RecN is required for survival during infection [35,36], suggesting they could be good drug targets.

Figure 3: Host-Pathogen drug filtering pipeline. This work-flow describes MTB drug target filtration process where the orange boxes represent the sets of unsuitable targets and the green are the suitable ones.
Next, we turned our attention to non-interacting targets. We were interested in the biological processes in which the 606 targets, which showed no evidence of interacting with human proteins, are involved in order to assess the potential biological roles in the organism. We first clustered the 400 annotated proteins out of these 606 targets using their functional similarity scores and left out proteins which were uncharacterized with respect to the GO biological process ontology to refine the statistical analysis results. Three clusters were found, as shown in Table 3, together with results of enrichment analysis of each cluster. These protein clusters are enriched in processes related to transcription, regulation and positive regulation of transcription, DNA-dependent, which is essential for responding to changing environments [37]. During infection, the host system triggers an immune response through the proinflammatory cytokine, allowing tumour necrosis factor alpha (TNF-α), together with interferon gamma (IFN-γ), to activate macrophages to produce nitric oxide synthase (NOS2) in order to kill intracellularly replicating MTB [38,39]. Here, we observed that some MTB protein clusters are enriched in response to nitrosative stress, indicating that these proteins will be active in a change in state or function of a cell in response to exposure to nitric oxide (NO). In order to ensure efficient uptake, MTB recruits a range of cell surface receptors in the host macrophage [40]. Proteins enriched in processes related to signal transduction and growth will enable bacterial pathogen cells to interpret, integrate and act upon external stimuli received by these cell surface receptors or by intracellular signals to achieve the desired cellular response. This indicates that this set of proteins is crucial for microbial pathogen survival and spreading strategies within the host for a long time at various stages of infection.
| Cluster | Proteins | Enriched Terms | GO Ids | GO term | Level | p-value | Bonferonni-correction |
|---|---|---|---|---|---|---|---|
| 1 | 216 | 2 | GO:0006351 | transcription, DNA-dependent | 8 | 2.18796×10−10 | 2.88811×10−08 |
| GO:0006355 | regulation of transcription,DNA-dependent | 8 | 1.59921×10−06 | 0.00021 | |||
| 2 | 120 | 5 | GO:0006355 | regulation of transcription,DNA-dependent | 8 | 1.84432×10−08 | 3.68863×10−06 |
| GO:0000160 | phosphorelay signal transductionsystem | 5 | 1.9038×10−07 | 3.80761×10−05 | |||
| GO:0035556 | intracellular signal transduction | 5 | 3.52753×10−07 | 7.05507×10−05 | |||
| GO:0006351 | transcription, DNA-dependent | 8 | 3.00490×10−06 | 0.00060 | |||
| GO:0051409 | response to nitrosative stress | 3 | 0.00016 | 0.03255 | |||
| 3 | 64 | 4 | GO:0040007 | growth | 1 | 2.01816×10−12 | 2.56307×10−10 |
| GO:0044119 | growth of symbiont in host cell | 7 | 2.50787×10−06 | 0.00032 | |||
| GO:0009097 | isoleucine biosynthetic process | 10 | 5.40389×10−05 | 0.00686 | |||
| GO:0045893 | positive regulation of transcription,DNA-dependent | 9 | 0.00039 | 0.04926 |
Table 3: Clustering MTB annotated target proteins that showed no evidence to interact with human proteins. Different clusters identified and associated statistically relevant GO biological process terms associated with the gene list in each cluster.
We used the MTB functional network to analyze paralogs of drug targets in terms of shared neighbors and whether these paralogs share similar network topological properties with the targets. 1783 MTB proteins have paralogs in the proteome, and among these proteins, 194 were in the list of 400 annotated protein targets. We computed the topological similarity between these target proteins and their paralogs in terms of number of shared neighbours using Jaccard index [41], given by
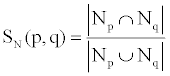 (2)
(2)
where |Nr| represents the number of protein neighbours to protein r in the functional network. Topological similarity scores between these target proteins and their paralogs showed a clear tendency to a high number of shared neighbors between protein targets and their paralogs, sharing 51.3% of neighbors, i.e., more than half identical neighbours, on average. Looking at the topological properties of these paralogs, we found that 124 out 194 protein targets have paralogs which are also targets or key proteins. This provides evidence that proteins playing a vital role in the system may have copies with the same characteristics for the survival of the system in the case of perturbations. As a consequence, a knockout or knockdown of such a target will be compensated for by its paralogs so that the system perturbation is negligible or less than expected. The extent of the observed similarity in the network patterns between protein targets and their paralogs also warrants further investigation in terms of their effects as targets.
The target proteins in the third cluster are involved in growth processes, indicating that these targets are essential for the growth of MTB in the host system. Since an efficient drug target should prevent growth of the pathogen, we identified which of our targets were essential for growth. 79 and 21 out of 400 annotated protein targets were found in the two lists of genes, which were experimentally identified by Sassetti et al. [35,36] to be required for normal MTB growth and for its survival during infection, respectively. Among these targets, 53 out of 79 targets required for MTB growth and 8 out of 21 targets required for its survival have no conserved paralogs within the pathogen. These proteins can be considered attractive and suitable targets for the discovery or rational design of novel anti-tubercular compounds, as they have been shown experimentally to be required for the functioning of organism.
We then looked at the total set of 606 proteins with no direct interaction with human proteins to find those with an indirect interaction. In general, the connectivity analysis of these targets reveals that most of the targets (522 out of 606) which do not interact with human proteins are direct neighbors of the 275 protein targets predicted to interact with human proteins. The remaining 84 targets that are not directly connected to targets interacting with human proteins have the following Tuberculosis functional classes (http://genolist.pasteur.fr/Tuberculist) assigned to them: 1 protein is involved in lipid metabolism, 8 in intermediary metabolism and respiration, 6 in virulence, detoxification and adaptation, 9 in cell wall and cell processes, 13 are regulatory proteins, 13 are PE/PPE proteins and 34 are still unknown or uncharacterized.
We looked at the predicted functional classes of the 34 unknown proteins and 13 proteins belonging to PE/PPE functional class. Of 34 unknown proteins, functional classes of 33 proteins were predicted, with 19 involved in cell wall and cell processes, 8 in intermediary metabolism and respiration, 3 are regulatory proteins, one is involved in virulence, detoxification and adaptation, one in lipid metabolism and one in insertion seqs and phages. For PE/PPE proteins, functional classes of 12 proteins were predicted 10 of which are involved in cell wall and cell processes, and 2 in intermediary metabolism and respiration. In fact, most of proteins of unknown and PE/PPE classes are predicted to be involved in cell wall and cell processes. It is known that the cell wall of MTB with its unusually low permeability plays a key role in its virulence, contributes crucially to the persistence of the pathogen in the host and is thought to contribute to the intrinsic drug resistance of mycobacteria [13]. This indicates that these proteins are likely to be important for the specific lifestyle of the organ ism and adaptability of this pathogen in the host, and thus may be appropriate candidate drug targets.
Finally, we put the 84 proteins through a number of other screens to determine their suitability as drug targets. We tried to assess this list by looking at UniProt predicted targets and “validated” drug targets in MTB on the TDR targets website (http://tdrtargets.org/). These candidate drug tar-gets include 8 genes which were in the UniProt target list and 1 in TDR validated targets. Among these 84 MTB protein targets which showed no direct connection to human proteins, 57 were observed to have no paralogs within the pathogen. 3 proteins out of these 57 protein targets were found to be essential for MTB growth and one is required for its survival during infection. These 3 proteins are Probable conserved membrane protein Rv0227c or MT0237 (P96409), involved in cell wall and cell processes, L-lysine 6-monooxygenase mbtG (O05820) suspected to be involved in lipid metabolism and the uncharacterized protein Rv0102/MT0111 (P64689) predicted to be involved in cell wall and cell processes. The putative un-characterized protein MT0185 (Q7DAC1) is required for MTB survival during infection. Interestingly, the 3 genes Rv0227c, mbtG and Rv0102 were previously identified as targets for drug development. The gene Rv0227c was qualified as a plausible target for drug design [42], mbtG was identified as an iron acquisition-related target [43], crucial for the survival within the host, and Rv0102 was identified to contribute actively to the MTB infection outcome [44].
Significant progress has been made in controlling TB using existing anti-tubercular drugs administrated for at least six months. This long duration of treatment contributes to the development of resistance and adding adverse effects of these drugs makes them prone to patient non-compliance. Further-more, as no new anti-tubercular drugs have been developed for over 20 years, it is increasingly important to pursue new and effective strategies to confront the challenge of TB in this 21st century. There is a need for the identification of novel and suitable drug targets within the bacterial pathogen that consider the host system in order to develop novel and effective therapeutics with anti-TB activity. However, the identification of novel drug targets for diseases and development of new drugs have been expensive and time-consuming, necessitating rational approaches and inexpensive technologies that shorten the length of time spent in drug discovery.
Understanding the host system and the complex interplay between pathogen and host may significantly enhance the identification of drug targets as these interactions are key factors in determining the outcome of the infection. Here, we used a map of protein-protein functional interactions between the human host and MTB, causative agent of TB. This map was obtained by integrating different genomic scale and microarray information and explored to filter the set of 881 protein targets previously identified within the MTB protein-protein functional network generated using genomics and functional data. We explored the human-MTB protein-protein functional interaction map to elucidate targets that also consider the host system to prevent potential adverse reactions in the host. The flowchart in Figure 3 summarizes MTB drug target filtration process, and the list of suitable targets (green boxes in the Figure) is in supplementary data. This has provided the opportunity to uncover putative protein targets for the development of novel and efficient anti-tubercular therapeutics to treat the disease.
A total of 606 targets out of 881 have shown no evidence of interacting directly with human proteins. We performed functional and statistical analyses in which uncharacterized proteins were removed, resulting in a list of 400 protein targets with functional annotations and no functional connections with human proteins. The biological process analysis of these targets have suggested that these proteins are likely to be important for the intracellular lifestyle of MTB, its adaptability and survival in the host by allowing the pathogen to acquire nutrients and to modulate the host response. No paralogs were observed within MTB for 206 out of 400 putative targets. We further filtered the possible targets based on other criteria, including essentiality for MTB growth and survival during infection. We have a final list of 67 potentially suitable candidates, some of which were previously identified. Novel identified targets and their suitability are results o f the integrative approach used through human or host, MTB and Human-MTB functional networks, and other functional analyses performed. This model provides a simple framework that can be used in drug target identification in order to produce a list of putative targets very rapidly at low cost.
NJM conceived the study, supervised and provided support on all aspects of the study. GKM performs all analysis and programming tasks, and wrote the paper. HAR contributed helpful suggestions for the analysis. All authors read and approved the final manuscript and NJM approved the production of the paper.
The authors especially wish to thank authors of freely available biological databases for making their data available to the scientific community. This work has been supported by the Claude Leon Foundation Postdoctoral Fellowship and the National Research Foundation (NRF) in South Africa through the Computational Biology (CBIO) research group at the Institute of Infectious Disease and Molecular Medicine, University of Cape Town.