Journal of Proteomics & Bioinformatics
Open Access
ISSN: 0974-276X
ISSN: 0974-276X
Research Article - (2017) Volume 10, Issue 11
Background: The relationships among phenotypes, genes, and variants play a key role for monogenic disorders in the era of precision medicine. Information about this is erupting with the current rapid development of genomic technology. However, it is time-consuming and error-prone to manually capture the information from the literature. Thus, how to capture this information rapidly and accurately is a bottleneck to be solved.
Results: Here, we present VarfromPDB, an automated and integrated method to mine the genes and variants related to a Mendelian disorder from multiple public curated databases and literature. To demonstrate the procedure, feasibility and application, we used a monogenic disorder, Joubert syndrome, as an example to capture the related genes from multiple sources including HPO, OrphaNet, ClinVar, UniProt and PubMed abstracts. The captured gene list is more comprehensive than that from DisGeNET and DISEASES databases.
Conclusion: VarfromPDB is a an automated and integrated tool to compile the up-to-date disease-genevariant database with comprehensive. It is valuable for genetic researchers and has great potential in facilitating the application of genetic testing for precision medicine. The source code for VarfromPDB is freely available at https://CRAN.R-project.org/package=VarfromPDB.
Keywords: Disease-gene-variant relations; Public databases; Mendelian disorder; Text mining; Automation; Bioinformatics
The rapid development of human genomics [1-4], disease genomics [5,6], and pharmacogenomics [7,8] brings a huge medical revolution with new patterns of health management, prevention, diagnosis, and treatment of diseases, and an era of personalized or precision medicine. In the precision medicine era, genetic testing becomes necessary for research, diagnosis, treatment and prognosis of diseases especially Mendelian disorders. Targeted sequencing and analyses are commonly employed, and many genetic testing products based on the targeted sequencing will be developed in the next few years. The phenotype-gene- variant database for a special Mendelian disorder or phenotype needs to be compiled for the product development and genetic research. Firstly, in the design stage of a study, the targeted regions and pathogenic variants need to be well understood. The challenge comes from deciphering genes, identifying causative mutations, and detecting disease-related genes because of genetic heterogeneity of Mendelian disorders. In general, there are dozens of causing genes for many Mendelian diseases. Secondly, the clinical significance of the detected variants also needs to be evaluated based on the population level and other evidences in the public databases and the literature. The information of phenotype-gene-variant relationships is continually increasing in the public databases and the literature. Thus, concurrent updates of the phenotype-gene-variant databases are essential.
Fortunately, some databases focusing on the relationships among human variants/genes and phenotypes are public and freely accessible. These include Human Phenotype Ontology (HPO), Orphanet, Online Mendelian Inheritance in Man (OMIM), ClinVar, and Universal Protein Resource (UniProt) etc. HPO provides a standardized vocabulary of phenotypic abnormalities encountered in human disease. HPO currently contains approximately 11,000 terms and over 115,000 annotations of genes linked to hereditary [9]. Orphanet is the reference portal for information on rare diseases and orphan drugs, and its aim is to help improve the diagnosis, care, and treatment of patients with rare diseases [10,11]. OMIM is a continuously updated catalog of human genes and genetic disorders and traits, with particular focus on the molecular relationship between genetic variation and phenotypic expression [12]. ClinVar is a freely accessible, public archive of reports of the relationships among human variations and phenotypes, with supporting evidence [13,14]. UniProt focuses on amino acid altering variants imported from Ensembl Variation databases. Variants that include human polymorphisms and disease mutations in the UniProt are manually curated from UniProtKB/Swiss-Prot [15].
These public databases collect not only disease-gene-variant relations, but also the names and aliases of diseases and clinical features, genes and variants. The databases may be compiled from the literature and other databases automatically, even manually, or submitted by the researchers directly and updated regularly. However, the information of a disorder may be not comprehensive for each database alone.
Additionally, PubMed provides the primary and latest source of the information. However, it is time-consuming and error-prone to capture the information from the databases one by one, and even to manually parse and search for information from the literature directly. Thus, how to capture the information rapidly, automatically and accurately is a bottleneck to be solved. Here, we present an automated and integrated tool to capture the genes and variants related to a Mendelian disorder from the public databases and PubMed abstracts. All the steps are realized in an easy-to-use R VarfromPDB package. Our package and the user manual are freely available at https://c-ran.rproject. org/web/packages/VarfromPDB/index.html.
The VarfromPDB package captures the genes and variants related to a Mendelian disorder from the public databases and PubMed abstracts. Below, we outline the main functionalities of the package. Figure 1 shows the design frame and data workflow in VarfromPDB.

Figure 1: The design frame and data workflow in VarfromPDB. The phenotype and alias were retrieved in HPO firstly, then genes and variants were captured in HPO, Orphanet, OMIM, ClinVar, UniProt and PubMed one by one. The genes and variants were compiled into a phenotype-gene-variant database based on the approved gene symbols in HGNC and variant nomenclature in HGVS.
Localize the public databases
The function localPDB performs the localization of the necessary files (Table 1) from several databases, including HPO, HGNC, Orphanet, OMIM, ClinVar, UniProt and UCSC. All the files can be freely accessed except for those from OMIM. An OMIM account and an API key should be applied for from OMIM website in advance. Each database can be specified flexibly, which can be selected depending on the database update frequency. The function download.file of R utils package was employed to download the files from the Internet.
| Database | File | Description | url |
|---|---|---|---|
| HPO | phenotype_annotation.tab | Clinical annotations | http://compbio.charite.de/hudson/job/hpo.annotations/lastStableBuild/artifact/misc/phenotype_annotation.tab |
| diseases_to_genes.txt | Links between genes and HPO-terms | http://compbio.charite.de/hudson/job/hpo.annotations.monthly/lastStableBuild/artifact/annotation/diseases_to_genes.txt | |
| HGNC | hgnc_complete_set.txt.gz | Complete HGNC dataset | ftp://ftp.ebi.ac.uk/pub/databases/genenames/hgnc_complete_set.txt.gz |
| ClinVar | variant_summary.txt.gz | All the annotations of variants submitted to ClinVar | ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/tab_delimited/variant_summary.txt.gz |
| gene_condition_source_id | report gene-disease relationships used in ClinVar, Gene, GTR and MedGen | ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/gene_condition_source_id | |
| OMIM | Morbidmap.txt | OMIM's Synopsis of the Human Gene Map | ftp://ftp.omim.org/OMIM/morbidmap |
| API | The similar information as the website but the robot is permitted | api.omim.org | |
| Uniprot | humsavar.txt | Index of manually curated Human polymorphisms and disease mutations from UniProtKB/Swiss-Prot. | ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/variants/humsavar.txt |
| Orphanet | en_product6.xml | Rare diseases with their associated genes | http://www.orphadata.org/data/xml/en_product6.xml |
| UCSC | refFlat.txt | The information of gene position and exons | http://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/refFlat.txt.gz |
Table 1: The necessary files from the public databases. These files are freely available from public databases except OMIM.
Get the aliases of a genetic disease
The function pheno_extract_HPO obtains the aliases of a genetic disease from HPO database for the given keyword(s), which can be a disease name or a clinical feature. The IDs of OMIM and Orphanet databases are captured based on the keyword(s) from the file ‘phenotype_annotation.tab’ (Table 1). The file provides the clinical annotations for each disease. All the records that contain the keyword(s) will be returned. When the keywords contain multiple words, the order of the multiple words will be ignored. The aliases of a genetic disease can be used in the capturing process from other databases to make sure the information as possible as comprehensive.
Capture the genes and variants relevant to a genetic disease/ phenotype from the public databases
The information on the relationship among genes and phenotypes for the given keyword(s) are extracted from several public databases including HPO, Orphanet, OMIM, ClinVar and UniProt. The gene names are transformed into approved symbols based on the HGNC database. Variants of the candidate genes are identified, and these may be the genes of interest. The associated phenotypes are verified whether they are related to the disease of interest or clinical feature. This process is carried out using 5 main functions including pheno_extract_HPO, extract_genes_orphanet, extract_omim, extract_clinvar and extract_ UniProt for each database.
Only the genes related to the keyword(s) can be extracted from the file ‘diseases_to_genes.txt’ (Table 1) in HPO, and the process is integrated into the function pheno_extract_HPO.
The function extract_genes_orphanet extracts only the relevant genes from the file ‘en_product6.xml’ (Table 1) in Orphanet database. The XML file is resolved depending on the R XML package. The function xmlTreeParse parses the XML file and generates internal nodes. Then the function getNodeSet finds XML nodes that match the string expression of ‘//Disorder’, and the function nodesToList in R XML2R package coerces XML nodes into a list. If the phenotype in a component of the list matches the keyword(s) or OrphanetID from pheno_extract_HPO, the component will be selected to extract the genes.
The function extract_omim extracts genes and variants from the OMIM database. The genes are extracted by searching for specific keyword(s) in the phenotypes in the file ‘morbidmap.txt’. The genes are checked, and the names are converted into approved symbols based on the HGNC summary file ‘hgnc_complete_set.txt’. If there are known variants of a gene, those will be captured from OMIM API, and this is needed to obtain an API key. The XML information from OMIM API is resolved similarly to that of the function extract_genes_orphanet, but the difference is start symbol of the nodes, the string expression of ‘//entry’ here. All the variants of the gene, the related phenotypes, and inheritance information are captured altogether. The genes from HPO and Orphanet or other information of interest can be added into the function.
The function extract_clinvar extracts the genes and variants from ClinVar database. The genes are extracted by searching for keyword(s) under the disease information in the file ‘gene_condition_source_id’. Then all the variants in the captured genes and the genes from HPO and Orphanet or other information of interest can be obtained from the file ‘variant_summary.txt’.
The function extract_UniProt extracts the genes and variants from the file ‘humsavar.txt’ in UniProt database, which focuses on the amino acid-altering variants manually curated human polymorphisms and disease mutations from UniProtKB/Swiss-Prot. The variants in the genes from HPO and Orphanet or other information of interest can also be added in the function too.
Capture the genes and variants from PubMed
The function extract_PubMed performs an equery in PubMed E-utilities using the search strategy similar to that of the web, and then captures the information from disease-related abstracts based on text mining. The functions in R RISmed package are employed to download and resolved the abstracts. The information of phenotypes, genes, variants, article titles, first authors, PubMed IDs, publication years, and publication journals will be captured.
In the text mining process of the phenotype information, a part of an abstract is split into multiple phrases by the prepositions and conjunctions, such as ‘due,’ ‘to,’ ‘by,’ ‘was,’ ‘were,’ ‘that,’ ‘of,’ ’in,’ ‘on,’ ‘is,’ ‘are,’ ‘the,’ ‘a,’ ‘an,’ ‘for,’ etc. The phenotype information can be identified in titles and conclusions by anchoring the keywords and high frequently used words such as ‘syndrome,’ ‘with,’ ‘Y-linked,’ ‘autosomal dominant,’ ‘cause*,’ ‘associated,’ etc. The high-frequency words are counted in the file ‘Morbidmap.txt’ in OMIM (Figure 2).

Figure 2: The high frequency words. The frequency is calculated from the phenotypes of OMIM.
The genes were extracted based on a dictionary-based method. An abstract is split into multiple words by the separators such as blank space, prepositions, conjunctions, or articles in the first. Then gene symbols and gene aliases in HGNC summary file can be captured.
To identify mutations, mutation nomenclature recommendations at the DNA level and protein-level followed by HGVS are searched for by regular expression and the names of amino acids. The gene names are checked and transformed into approved symbols. The protein changes are transformed into 3-character abbreviations of amino acids.
When there are multiple genes and variants reported in one article, each gene-variant relationship pairs need to be resolved one by one based on the bipartite graph theory and sentence-level concurrence. Let G = (U, V, E) denote a bipartite graph (Figure 3), which consists of 2 disjoint sets (U and V) and edges E. Every edge connects a vertex in U to one in V.

Figure 3: Three cases for resolving multiple genes and variants. (1) A complete bipartite graph Km, n has m*n edges. Each edge represents a potential relation pair to be resolved. (2) All the vertexes can be covered by the available pairs. (3) One or more vertexes in either of the 2 disjoint sets (U and V) cannot be covered by the available pair(s). (4) One or more vertexes in both of the 2 disjoint sets (U and V) cannot be covered by the available pair(s).
Supposed |U| = m and |V| = n, then for a complete bipartite graph Km, n, there are m*n edges, and the size of edge cover is equal to max(m, n).
Suppose U and V represents several genes and variants in an abstract, respectively, with E denoting the gene-variant relationship pairs. Then the size of relationship pairs is not more than the size of edge cover, because there is no available gene-variant pair for some vertexes in some cases when a gene and a variant do not occur in one sentence. Some vertexes represent the genes and variants here.
All the edges are checked whether it follows the concurrence at sentence level or phrase level, which means 2 vertexes of an edge should occur in one sentence or a phrase. A paragraph should be split by the separators including comma, period, and the word ‘and.’ If there are multiple possible edges for a vertex, the priority of the separators should be considered. The comma has the top priority. Only the edges with vertexes following the concurrence will be retained. (Figure 3) shows 3 cases for resolving multiple genes and variants.
Compile the genes and variants
The function genes compile compiles the gene sets from different databases to generate a union set according to the approved gene symbols, and then the related phenotypes from different databases and physical positions of the genes are annotated. The physical positions are extracted from the UCSC file ‘refFlat.txt’ (Table 1). The function genes_add_PubMed compares the genes with reported variants from PubMed abstracts with that from the public databases, and then the additional gene-phenotype pairs are added.
In order to rank the captured relations, a VarfromPDB score that ranging from 0 to 1 is computed based on the evidences from the curated databases and literature as follows:
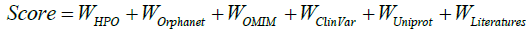
Where
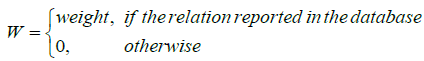
All the weights are set 0.2 for all the databases except for 0.1 of HPO, and
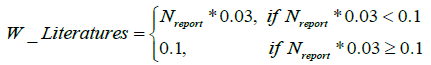
For the weight of the evidences from literature, each report give the evidence score 0.03. The function variants compile compiles a union of the variants from different databases. The variants are compared between ClinVar and other databases. Firstly, the captured variants from OMIM are compared with ClinVar by the OMIM variants ID, and that from UniProt is compared with ClinVar by the protein changes. Finally, a union of phenotype-gene-variant relationships with the ClinVar-like format can be obtained consisting of the additional variants and the set from ClinVar.
To demonstrate the procedure, feasibility and application, we used a monogenic disorder, Joubert syndrome, as an example to capture the related genes by VarfromPDB package. We automatically captured the genes and variants related to Joubert syndrome from the public databases and PubMed abstracts using the R script (Additional file 2_VarfromPDB_Joubert.r). The script is consisted of the functions of VarfromPDB package with “Joubert syndrome” as the keyword. More detailed descriptions of all the functions can be seen in the package manual and vignettes (http://rpubs.com/Zongfu/270012). The phenotype-gene-variant relationships are automatically filtered by the gene status: 1) have an approved symbol; 2) with definite physical positions; 3) with more than one definite mutation on the gene. Overall, there are 36 genes related to Joubert syndrome (Additional file 1: Table S1) captured from public databases and PubMed abstracts using VarfromPDB package. The genes are in descending order by evidence score.
In order to evaluate the comprehensiveness of the gene list, we got that with “Joubert syndrome” as the keyword from two published websites including DisGeNET [16,17] and DISEASES [18], both of which are comprehensive platform integrating information on human disease-associated genes and variants. We got 19 and 15 genes related to Joubert syndrome from DisGeNET and DISEASES, respectively.
All the gene lists were compared and manually checked. We found that the genes related to Joubert syndrome using VarfromPDB package are more comprehensive than that from other methods. All the 19 genes from DisGeNET and 15 genes from DISEASES can be captured by VarfromPDB (Figure 4). There are additional 15 genes related to Joubert syndrome and two false positive genes including NEB and PDE7B after manually checking (labelled in the column ‘comparison’ of Additional file 1). Two causing genes including POC1B and INVS captured from PubMed abstracts, have not been enrolled in the several databases yet.

Figure 4: Comparison between different gene lists. VarfromPDB can capture more comprehensive genes related to Joubert syndrome, which can capture not only the 19 and 15 genes related to Joubert syndrome from DisGeNET and DISEASES, but also additional 15 genes.
It needs about ten minutes to extract and integrate the information from the local database. However, the run time of localPDB step depends on internet speed.
We here present VarfromPDB, a bioinformatics tool to capture the genes and variants from the public databases including HPO, Orphanet, OMIM, ClinVar, UniProt and PubMed abstracts. VarfromPDB just takes the keyword(s) as input for a given genetic disorder. VarfromPDB considers the aliases of a disease and a gene, and integrate the phenotype-gene-variant relationships automatically. The process is implemented in an easy-to-use and fully documented R package that describes each step in detail.
The genes related to a genetic disorder are comprehensive and can be well captured from the public databases and PubMed abstracts. By analyses of the genes related to Joubert syndrome, we demonstrated that VarfromPDB can dig more comprehensive genes than DisGeNET and DISEASES databases. VarfromPDB captured not only all the 19 and 15 genes related to Joubert syndrome from DisGeNET and DISEASES, but also additional 15 Joubert syndrome related genes.
This approach is more efficient and accurate than the traditional strategy. It is time-consuming and error-prone using the traditional strategy, because there are too many reports to read to get the information of genotypes and phenotypes manually. In contrast, our approach is automated to mine the information from PubMed abstracts and public databases including HPO, Orphanet, OMIM, ClinVar and UniProt.
However, we also observed the genes related to a genetic disorder only in public databases may be incomplete. Two of the 36 Joubert syndrome related genes was missed in the public databases. One reason is the lag on updates. There may be almost several weeks, even months, and later than the time of the publication. The second reason is that many gene names are easily confusing. For example, MRI can be a gene symbol alias; meanwhile, it can represent a medical imaging technique ‘Magnetic resonance imaging’. It is a very big challenge to capture the genes from the abstracts, although we have a definite set of gene symbols and aliases from HGNC. Thus, we had to manually check the results carefully. Another reason is the issue on the variants. Although HGVS have provided the clear nomenclature recommendation for many years, some variants nomenclatures in the PubMed reports do not follow the recommendation, especially in the historical literature. Some variants were not mentioned in the abstracts for some reason, so mining for variants in the full text may be necessary in the future.
Note that Manual checking is the last but very important step in the process, especially for the information captured from PubMed abstracts considering the above challenges. We should pay more attention to the additional genes with the score less than 0.1, which are usually missed in the public databases.
Some variants in public databases may be submitted directly by certain organizations or labs and never reported in PubMed, so these variants may be missed from PubMed. Therefore, the information from either public databases or PubMed abstracts may be incomplete. The recommended strategy is to combine the information from different sources.
VarfromPDB can well capture the genes and variants related to a genetic, especially Mendelian disorder, from public databases and literature from PubMed with comprehensive, efficient and automated. Therefore, VarfromPDB is valuable for genetic researchers and has great potential for facilitating the application of genetic testing in the precision medicine.
Note that the current version of VarfromPDB has two limitations to be addressed in future. One is the genetic heterogeneity in different global populations. We are trying to find or compile a dictionary of the region and population information to mine in PubMed. The second is the mining extent. We plan to perform text mining in the full articles rather than the abstracts. These limitations will all be addressed in the next few months. A friendly web-server for the pipeline has been developed, and will be public soon.